WO2016094420A1 - Non-coding rnas and uses thereof - Google Patents
Non-coding rnas and uses thereof Download PDFInfo
- Publication number
- WO2016094420A1 WO2016094420A1 PCT/US2015/064525 US2015064525W WO2016094420A1 WO 2016094420 A1 WO2016094420 A1 WO 2016094420A1 US 2015064525 W US2015064525 W US 2015064525W WO 2016094420 A1 WO2016094420 A1 WO 2016094420A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- lncrna
- expression
- coding
- transcripts
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
Definitions
- compositions and methods for cancer diagnosis, research and therapy including but not limited to, cancer markers.
- cancer markers include but not limited to, cancer markers.
- non-coding RNAs as diagnostic markers and clinical targets for cancer.
- prostate cancer is a leading cause of male cancer-related death, second only to lung cancer (Abate-Shen and Shen, Genes Dev 14:2410 [2000]; Ruijter et al , Endocr Rev, 20:22 [1999]).
- the American Cancer Society estimates that about 184,500 American men will be diagnosed with prostate cancer and 39,200 will die in 2001.
- Prostate cancer is typically diagnosed with a digital rectal exam and/or prostate specific antigen (PSA) screening.
- PSA prostate specific antigen
- An elevated serum PSA level can indicate the presence of PCA.
- PSA is used as a marker for prostate cancer because it is secreted only by prostate cells.
- a healthy prostate will produce a stable amount ⁇ typically below 4 nanograms per milliliter, or a PSA reading of "4" or less ⁇ whereas cancer cells produce escalating amounts that correspond with the severity of the cancer.
- a level between 4 and 10 may raise a doctor's suspicion that a patient has prostate cancer, while amounts above 50 may show that the tumor has spread elsewhere in the body.
- a transrectal ultrasound is used to map the prostate and show any suspicious areas.
- Biopsies of various sectors of the prostate are used to determine if prostate cancer is present.
- Treatment options depend on the stage of the cancer. Men with a 10-year life expectancy or less who have a low Gleason number and whose tumor has not spread beyond the prostate are often treated with watchful waiting (no treatment).
- Treatment options for more aggressive cancers include surgical treatments such as radical prostatectomy (RP), in which the prostate is completely removed (with or without nerve sparing techniques) and radiation, applied through an external beam that directs the dose to the prostate from outside the body or via low-dose radioactive seeds that are implanted within the prostate to kill cancer cells locally.
- RP radical prostatectomy
- radiation applied through an external beam that directs the dose to the prostate from outside the body or via low-dose radioactive seeds that are implanted within the prostate to kill cancer cells locally.
- PSA prostate specific antigen
- compositions and methods for cancer diagnosis, research and therapy including but not limited to, cancer markers.
- cancer markers include but not limited to, cancer markers.
- non-coding RNAs as diagnostic markers and clinical targets for cancer.
- the present disclosure provides a method of screening for the presence of cancer in a subject, comprising (a) contacting a biological sample from a subject with a gene expression detection assay, wherein said gene expression detection assay comprises a gene expression informative reagent for identification of the level of expression of one or more non- coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309; (b) detecting the level of expression of said non-coding in said sample using an in vitro assay; and (c) diagnosing cancer in said subject when an increased level of expression of said non-coding RNAs in said sample relative to the level in normal cells is detected.
- the RNAs are converted to cDNA prior to or during detection.
- the sample is selected from, for example, tissue, blood, plasma, serum, urine, urine supernatant, urine cell pellet, semen, prostatic secretions or prostate cells.
- the detection is carried out utilizing a method selected from, for example, a sequencing technique, a nucleic acid hybridization technique, or a nucleic acid amplification technique.
- the nucleic acid amplification technique is selected from, for example, polymerase chain reaction, reverse transcription polymerase chain reaction, transcription-mediated amplification, ligase chain reaction, strand displacement amplification, or nucleic acid sequence based amplification. The present disclosure is not limited to a particular cancer.
- the reagent is a pair of amplification oligonucleotides, a sequencing primer, or an oligonucleotide probe.
- the reagent comprises one or more labels.
- the one or more non-coding RNAs is two or more (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 50, 75, 100, or more).
- a probe set for assessing a cancer status of a subject comprising a plurality of probes, wherein the probes in the probe set are capable of detecting an expression level of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1- 2309 or the corresponding cDNA.
- compositions comprising one or more reaction mixtures, wherein each reaction mixture comprises a complex of a non-coding RNAs selected from those described by SEQ ID NOs: 1-2309 or the corresponding cDNA and a probe that binds to said non- coding RNA.
- Figure 1 shows that ⁇ 4Z> initio transcriptome assembly reveals an expansive landscape of human transcription
- Figure 2 shows characterization of the MiTranscriptome assembly,
- (a) Pie chart of composition and quantities of IncRNA, transcripts of unknown coding potential (TUCP), expressed pseudogene, read-through, and protein-coding genes in the MiTranscriptome assembly (b) Pie charts of number of IncRNAs and TUCP genes (top) unannotated versus annotated relative to reference catalogs and (bottom) intragenic versus intergenic.
- Figure 4 shows discovery of lineage-associated and cancer-associated IncRNAs in the MiTranscriptome compendia, (a) Heatmap of lineage-specific IncRNAs. (b) Heatmap of cancer- specific IncRNAs nominated by SSEA Cancer vs. Normal analysis of 12 cancer types (columns), (c) Scatter plots showing enrichment score for Cancer vs.
- Figure 5 shows curation and processing of samples in the MiTrascriptome compendia.
- Figure 7 shows meta assembly, a, schematic of transcriptome meta-assembly algorithm using a simplified example with three transtrags transcribed from left to right, b, The pruned splice graph from panel a is subjected to meta-assembly.
- c Genome view showing an example of the meta- assembly procedure for breast cohort transfrags in a chromosome 12ql3.3 locus containing the LncRNA HOTAIR and the protein-coding gene HOXC11 on opposite strands (chrl2:54,349,995- 54,377,376, hgl9).
- Figure 8 shows characterization of unannotated transcripts
- a Bar plots comparing numbers of unannolaled versus different chipses of annotated transcripts for each of the 18 cohorts
- b 001 plots depicting comparison of MiTransriptome with reference transcripts from RefSeq, UCSC, or GENCODE.
- c Dot plots comparing the basewise, splice site, and splicing partem precision and sensitivity of MiTranscriptome and GENCODE using RefSeq (left) or Cabili et al. LncRNAs (right)
- Figure 9 shows classification of transcripts of unknown coding potential
- a Decision tree showing categorization of ab initio transcripts
- b ROC curve comparing false positive rate (x axis) with true positive rate (yaxis) for CPAT coding potential predictions of ncRNAs versus protein- coding genes
- c Curve comparing probability cutoff (x axis) with balanced accuracy (yaxis).
- d Scatter plot comparing frequencies of Pfam domain occurrences in non-transcribed intergenic space versus transcribed regions
- e Three-dimensional scatter plot comparing Fickett score (x axis), ORF size (yaxis), and Hexamer score (z axis) for all transcripts.
- f g,h Boxplots comparing ORF size (f), Hexamer score (g), and Fickett score (h).
- Figure 10 shows Mitranscriptome characterization, a, Comparison of the relationship of maximum number of exons per gene to the number of isoforms per gene, b, Density histogram depicting the confidence scores for annotated and unannotated IncRNAs. c, Cumulative distribulion plot for basewise conservation fraction of proteins, read-throughs, pseudogenes, TUCPs, IncRNAs. d, Bar plot showing KS test statistics for classes of transcripts versus random intergenic controls, e, Cumulative dislribution plot for promoter conservation (legend shared with a), f, Bar plot showing KS tests for promoter conservation versus random intergenic regions, g, ROC curve for predicting conservation of protein coding genes versus random intergenic controls.
- Figure 11 shows validation of lncRNA transcripts.
- Figure 12 shows validation of lncRNA transcripts, a, b, Representative example of two of twenty previously unannotated lncRNA transcripts that were analyzed by Sanger sequencing to ensure primer specificity with their associated chromatograms.
- c Heatmap representation of the correlation between qPCR (fold change over median) with RNA-seq (FPKM) of 100 selected transcripts in cell lines A549, LNCaP, and MCF7.
- Figure 13 shows enrichment of MiTranscriptome assembly for disease-associated regions, a
- MiTranscriptome assembly in comparison to reference catalog
- b Pie charts comparing distributions of intronic and exonic GWAS SNP coverage of the MiTranscriptome assembly (left) and reference catalogs (right)
- c Dot plot showing enrichment of GWAS SNPs (cirde) versus random SNPs (diamond) for novel intergenic IncRNAs and TUCPs.
- Figure 14 shows discovery of lineage associated and cancer associated transcripts, a, Heatmap of lineage-specific transcripts (LATs) nominated by SSEA. b, Heatmap of cancer-specific transcripts (CATS) nominated by SSEA.
- LATs lineage-specific transcripts
- CAS cancer-specific transcripts
- Figure 15 shows lineage-specific and cancer-specific transcripts, a, Scatter plot grid showing lineage-specific and cancer-specific transcripts (CLATs) nominated by SSEA. b and c, Boxplots comparing the perfonnance of (b) positively enriched CLATs and c) negatively enriched CLATs for each transcript category across 12 cancer types.
- Figure 16 shows examples of cancer and/or lineage associated transcripts), a, Genomic view of chromosome 6q26-q271ocus. b, Expression data for MEAT6 (demarcated by asterisk in a).
- the terms “detect”, “detecting” or “detection” may describe either the general act of discovering or discerning or the specific observation of a composition. Detecting a
- composition may comprise determining the presence or absence of a composition. Detecting may comprise quantifying a composition. For example, detecting comprises determining the expression level of a composition.
- the composition may comprise a nucleic acid molecule.
- the composition may comprise at least a portion of the ncRNAs disclosed herein. Alternatively, or additionally, the composition may be a detectably labeled composition.
- the term "subject” refers to any organisms that are screened using the diagnostic methods described herein. Such organisms preferably include, but are not limited to, mammals (e.g., murines, simians, equines, bovines, porcines, canines, felines, and the like), and most preferably includes humans. Alternatively, the organism is an avian, amphibian, reptile or fish.
- mammals e.g., murines, simians, equines, bovines, porcines, canines, felines, and the like
- the organism is an avian, amphibian, reptile or fish.
- diagnosis refers to the recognition of a disease by its signs and symptoms, or genetic analysis, pathological analysis, histological analysis, and the like.
- the term "characterizing cancer in a subject” refers to the identification of one or more properties of a cancer sample in a subject, including but not limited to, the presence of benign, pre-cancerous or cancerous tissue, the stage of the cancer, and the subject's prognosis.
- Cancers may be characterized by the identification of the expression of one or more cancer marker genes, including but not limited to, the ncRNAs disclosed herein.
- stage of cancer refers to a qualitative or quantitative assessment of the level of advancement of a cancer. Criteria used to determine the stage of a cancer include, but are not limited to, the size of the tumor and the extent of metastases (e.g., localized or distant).
- nucleic acid molecule refers to any nucleic acid containing molecule, including but not limited to, DNA or RNA.
- the nucleic acid molecule may comprise one or more nucleotides.
- the term encompasses sequences that include any of the known base analogs of DNA and RNA including, but not limited to, 4-acetylcytosine, 8-hydroxy-N6-methyladenosine, aziridinylcytosine, pseudoisocytosine, 5-(carboxyhydroxylmethyl) uracil, 5-fluorouracil,
- 5-methylaminomethyluracil 5-methoxyaminomethyl-2-thiouracil, beta-D-mannosylqueosine, 5'-methoxycarbonylmethyluracil, 5 -methoxy uracil, 2-methylthio-N6-isopentenyladenine, uracil-5-oxyacetic acid methylester, uracil-5-oxyacetic acid, oxybutoxosine, pseudouracil, queosine,
- 2- thiocytosine 5-methyl-2-thiouracil, 2-thiouracil, 4-thiouracil, 5-methyluracil, N-uracil-5-oxyacetic acid methylester, uracil-5-oxyacetic acid, pseudouracil, queosine, 2-thiocytosine, and
- gene refers to a nucleic acid (e.g. , DNA) sequence that comprises coding sequences necessary for the production of a polypeptide, precursor, or RNA (e.g., rRNA, tRNA).
- the polypeptide can be encoded by a full length coding sequence or by any portion of the coding sequence so long as the desired activity or functional properties (e.g., enzymatic activity, ligand binding, signal transduction, immunogenicity, etc.) of the full-length or fragments are retained.
- the term also encompasses the coding region of a structural gene and the sequences located adjacent to the coding region on both the 5' and 3' ends for a distance of about 1 kb or more on either end such that the gene corresponds to the length of the full-length mRNA. Sequences located 5' of the coding region and present on the mRNA are referred to as 5' non-translated sequences. Sequences located 3' or downstream of the coding region and present on the mRNA are referred to as 3' non-translated sequences.
- the term "gene” encompasses both cDNA and genomic forms of a gene.
- a genomic form or clone of a gene contains the coding region interrupted with non-coding sequences termed "introns” or “intervening regions” or “intervening sequences.”
- Introns are segments of a gene that are transcribed into nuclear RNA (hnRNA); introns may contain regulatory elements such as enhancers. Introns are removed or “spliced out” from the nuclear or primary transcript; introns therefore are absent in the messenger RNA (mRNA) transcript.
- mRNA messenger RNA
- oligonucleotide refers to a short length of single-stranded polynucleotide chain. Oligonucleotides are typically less than 200 residues long (e.g. , between 15 and 100), however, as used herein, the term is also intended to encompass longer polynucleotide chains. Oligonucleotides are often referred to by their length. For example a 24 residue
- Oligonucleotide is referred to as a "24-mer”. Oligonucleotides can form secondary and tertiary structures by self-hybridizing or by hybridizing to other polynucleotides. Such structures can include, but are not limited to, duplexes, hairpins, cruciforms, bends, and triplexes.
- the terms “complementary” or “complementarity” are used in reference to polynucleotides (i.e. , a sequence of nucleotides) related by the base-pairing rules.
- sequence “5'-A-G-T-3'” is complementary to the sequence “3'-T-C-A-5 ⁇ ”
- Complementarity may be “partial,” in which only some of the nucleic acids' bases are matched according to the base pairing rules. Or, there may be “complete” or “total” complementarity between the nucleic acids.
- the degree of complementarity between nucleic acid strands has significant effects on the efficiency and strength of hybridization between nucleic acid strands. This is of particular importance in amplification reactions, as well as detection methods that depend upon binding between nucleic acids.
- a partially complementary sequence is a nucleic acid molecule that at least partially inhibits a completely complementary nucleic acid molecule from hybridizing to a target nucleic acid is "substantially homologous.”
- the inhibition of hybridization of the completely complementary sequence to the target sequence may be examined using a hybridization assay (Southern or Northern blot, solution hybridization and the like) under conditions of low stringency.
- a substantially homologous sequence or probe will compete for and inhibit the binding (i.e. , the hybridization) of a completely homologous nucleic acid molecule to a target under conditions of low stringency.
- low stringency conditions require that the binding of two sequences to one another be a specific (i.e. , selective) interaction.
- the absence of non-specific binding may be tested by the use of a second target that is substantially non-complementary (e.g. , less than about
- hybridization is used in reference to the pairing of complementary nucleic acids. Hybridization and the strength of hybridization (i.e. , the strength of the association between the nucleic acids) is impacted by such factors as the degree of complementary between the nucleic acids, stringency of the conditions involved, the T m of the formed hybrid, and the G:C ratio within the nucleic acids. A single molecule that contains pairing of complementary nucleic acids within its structure is said to be “self-hybridized.”
- stringency is used in reference to the conditions of temperature, ionic strength, and the presence of other compounds such as organic solvents, under which nucleic acid hybridizations are conducted.
- low stringency conditions a nucleic acid sequence of interest will hybridize to its exact complement, sequences with single base mismatches, closely related sequences (e.g. , sequences with 90% or greater homology), and sequences having only partial homology (e.g. , sequences with 50-90% homology).
- 'medium stringency conditions a nucleic acid sequence of interest will hybridize only to its exact complement, sequences with single base mismatches, and closely relation sequences (e.g. , 90% or greater homology).
- a nucleic acid sequence of interest will hybridize only to its exact complement, and (depending on conditions such a temperature) sequences with single base mismatches. In other words, under conditions of high stringency the temperature can be raised so as to exclude hybridization to sequences with single base mismatches.
- isolated when used in relation to a nucleic acid, as in "an isolated
- oligonucleotide or "isolated polynucleotide” refers to a nucleic acid sequence that is identified and separated from at least one component or contaminant with which it is ordinarily associated in its natural source. Isolated nucleic acid is such present in a form or setting that is different from that in which it is found in nature. In contrast, non-isolated nucleic acids as nucleic acids such as DNA and RNA found in the state they exist in nature. For example, a given DNA sequence (e.g.
- RNA sequences such as a specific mRNA sequence encoding a specific protein, are found in the cell as a mixture with numerous other mRNAs that encode a multitude of proteins.
- isolated nucleic acid encoding a given protein includes, by way of example, such nucleic acid in cells ordinarily expressing the given protein where the nucleic acid is in a chromosomal location different from that of natural cells, or is otherwise flanked by a different nucleic acid sequence than that found in nature.
- the isolated nucleic acid, oligonucleotide, or polynucleotide may be present in single-stranded or double-stranded form.
- the oligonucleotide or polynucleotide When an isolated nucleic acid, oligonucleotide or polynucleotide is to be utilized to express a protein, the oligonucleotide or polynucleotide will contain at a minimum the sense or coding strand (i.e. , the oligonucleotide or polynucleotide may be single-stranded), but may contain both the sense and anti-sense strands (i.e. , the oligonucleotide or polynucleotide may be double-stranded).
- the term "purified” or "to purify” refers to the removal of components (e.g., contaminants) from a sample.
- label refers to any atom or molecule that can be used to provide a detectable (preferably quantifiable) effect, and that can be attached to a nucleic acid or protein. Labels include but are not limited to dyes; radiolabels such as 2 P; binding moieties such as biotin; haptens such as digoxgenin; luminogenic, phosphorescent or fluorogenic moieties; and fluorescent dyes alone or in combination with moieties that can suppress or shift emission spectra by
- Labels may provide signals detectable by fluorescence, radioactivity, colorimetry, gravimetry, X-ray diffraction or absorption, magnetism, enzymatic activity, and the like.
- a label may be a charged moiety (positive or negative charge) or alternatively, may be charge neutral.
- Labels can include or consist of nucleic acid or protein sequence, so long as the sequence comprising the label is detectable. In some embodiments, nucleic acids are detected directly without a label (e.g., directly reading a sequence).
- sample is used in its broadest sense. In one sense, it is meant to include a specimen or culture obtained from any source, as well as biological and environmental samples. Biological samples may be obtained from animals (including humans) and encompass fluids, solids, tissues, and gases. Biological samples include blood products, such as plasma, serum and the like. Such examples are not however to be construed as limiting the sample types applicable to the present disclosure.
- compositions and methods for cancer diagnosis, research and therapy including but not limited to, cancer markers.
- cancer markers include but not limited to, cancer markers.
- non-coding RNAs as diagnostic markers and clinical targets for cancer.
- RNA transcripts are not classical protein-coding genes. There is an abundance of unknown, uncharacterized RNA species in the human transcriptome (e.g., lncRNA or long non- coding RNAs). Provided herein are compositions and methods for utilizing such non-coding RNAs in diagnostic, research, and screening methods.
- RNAs include, but are not limited to, those described in SEQ ID NOs: 1-2309. Exemplary, non-limiting methods are described herein.
- the sample may be tissue (e.g. , a biopsy sample, a prostate biopsy sample or a tissue sample obtained by prostatectomy), blood, urine, semen, prostatic secretions or a fraction thereof (e.g., plasma, serum, urine supernatant, urine cell pellet, cells or prostate cells).
- a urine sample may be collected immediately following an attentive digital rectal examination (DRE), which causes prostate cells from the prostate gland to shed into the urinary tract.
- DRE attentive digital rectal examination
- the patient sample is subjected to preliminary processing designed to isolate or enrich the sample for the non-coding RNAs or cells that contain the non-coding RNAs.
- preliminary processing designed to isolate or enrich the sample for the non-coding RNAs or cells that contain the non-coding RNAs.
- a variety of techniques known to those of ordinary skill in the art may be used for this purpose, including but not limited to: centrifugation; immunocapture; cell lysis; nucleic acid amplification; and, nucleic acid target capture (See, e.g., EP Pat. No. 1 409 727, herein incorporated by reference in its entirety).
- the non-coding RNAs may be detected along with other markers in a multiplex or panel format. Markers may be selected for their predictive value alone or in combination with non-coding RNAs described herein (e.g., one or more of SEQ ID NOs: 1-2309).
- Exemplary prostate cancer markers include, but are not limited to: AMACR/P504S (U.S. Pat. No. 6,262,245); PCA3 (U.S. Pat. No. 7,008,765); PCGEMl (U.S. Pat. No. 6,828,429); prostein/P501 S, P503S, P504S, P509S, P510S, prostase/P703P, P710P (U.S. Publication No. 20030185830); RAS/KRAS (Bos, Cancer Res.
- multiplex or array formats are utilized to detect multiple markers in combination.
- the level of expression of one or more, 2 or more, 3 or more, 4 or more, 5 or more, 10 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more non-coding RNAs (ncRNAs) is utilized in the research, screening, diagnostic and prognositic compositions and methods described herein.
- the one or more ncRNAs may be selected from the group comprising. i. DNA and RNA Detection
- RNAs of the present disclosure are detected using a variety of nucleic acid techniques known to those of ordinary skill in the art, including but not limited to: nucleic acid sequencing; nucleic acid hybridization; and, nucleic acid amplification.
- the methods, compositions and kits may comprise one or more ncRNAs.
- the methods, compositions and kits may comprise 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 15 or more, 20 or more, 25 or more, 30 or more, 40 or more, 45 or more, 50 or more, 55 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more, 130 or more, 140 or more, 150 or more ncRNAs.
- the one or more ncRNAs may be selected from, for example, those described in SEQ ID NO: 1
- nucleic acid sequencing methods are utilized (e.g., for detection of amplified nucleic acids).
- the technology provided herein finds use in a Second Generation (a.k.a. Next Generation or Next-Gen), Third Generation (a.k.a. Next-Next-Gen), or Fourth Generation (a.k.a. N3-Gen) sequencing technology including, but not limited to, pyrosequencing, sequencing-by-ligation, single molecule sequencing, sequence-by-synthesis (SBS), semiconductor sequencing, massive parallel clonal, massive parallel single molecule SBS, massive parallel single molecule real-time, massive parallel single molecule real-time nanopore technology, etc.
- RNA is less stable in the cell and more prone to nuclease attack experimentally RNA is usually reverse transcribed to DNA before sequencing.
- DNA sequencing techniques are suitable, including fluorescence-based sequencing methodologies (See, e.g., Birren et al, Genome Analysis: Analyzing DNA, 1, Cold Spring Harbor, N.Y.; herein incorporated by reference in its entirety).
- fluorescence-based sequencing methodologies See, e.g., Birren et al, Genome Analysis: Analyzing DNA, 1, Cold Spring Harbor, N.Y.; herein incorporated by reference in its entirety.
- the technology finds use in automated sequencing techniques understood in that art.
- the present technology finds use in parallel sequencing of partitioned amplicons (PCT Publication No:
- the technology finds use in DNA sequencing by parallel oligonucleotide extension
- NGS Next-generation sequencing
- NGS methods can be broadly divided into those that typically use template amplification and those that do not.
- Amplification-requiring methods include pyrosequencing commercialized by Roche as the 454 technology platforms (e.g., GS 20 and GS FLX), Life Technologies/Ion Torrent, the Solexa platform commercialized by Illumina, GnuBio, and the Supported Oligonucleotide Ligation and Detection (SOLiD) platform commercialized by Applied Biosystems.
- Non-amplification approaches also known as single-molecule sequencing, are exemplified by the HeliScope platform commercialized by Helicos Biosciences, and emerging platforms commercialized by VisiGen, Oxford Nanopore Technologies Ltd., and Pacific Biosciences, respectively.
- template DNA is fragmented, end-repaired, ligated to adaptors, and clonally amplified in-situ by capturing single template molecules with beads bearing oligonucleotides complementary to the adaptors.
- Each bead bearing a single template type is compartmentalized into a water-in-oil microvesicle, and the template is clonally amplified using a technique referred to as emulsion PCR.
- the emulsion is disrupted after amplification and beads are deposited into individual wells of a picotitre plate functioning as a flow cell during the sequencing reactions. Ordered, iterative introduction of each of the four dNTP reagents occurs in the flow cell in the presence of sequencing enzymes and luminescent reporter such as luciferase.
- sequencing data are produced in the form of shorter-length reads.
- single-stranded fragmented DNA is end-repaired to generate 5'-phosphorylated blunt ends, followed by Klenow-mediated addition of a single A base to the 3' end of the fragments.
- Klenow-mediated addition facilitates addition of T- overhang adaptor oligonucleotides, which are subsequently used to capture the template-adaptor molecules on the surface of a flow cell that is studded with oligonucleotide anchors.
- the anchor is used as a PCR primer, but because of the length of the template and its proximity to other nearby anchor oligonucleotides, extension by PCR results in the "arching over" of the molecule to hybridize with an adjacent anchor oligonucleotide to form a bridge structure on the surface of the flow cell.
- These loops of DNA are denatured and cleaved. Forward strands are then sequenced with reversible dye terminators.
- the sequence of incorporated nucleotides is determined by detection of post- incorporation fluorescence, with each fluor and block removed prior to the next cycle of dNTP addition. Sequence read length ranges from 36 nucleotides to over 250 nucleotides, with overall output exceeding 1 billion nucleotide pairs per analytical run.
- interrogation probes In the SOLiD system, interrogation probes have 16 possible combinations of the two bases at the 3' end of each probe, and one of four fluors at the 5' end. Fluor color, and thus identity of each probe, corresponds to specified color-space coding schemes. Multiple rounds (usually 7) of probe annealing, ligation, and fluor detection are followed by denaturation, and then a second round of sequencing using a primer that is offset by one base relative to the initial primer. In this manner, the template sequence can be computationally reconstructed, and template bases are interrogated twice, resulting in increased accuracy. Sequence read length averages 35 nucleotides, and overall output exceeds 4 billion bases per sequencing run.
- the technology finds use in nanopore sequencing (see, e.g., Astier et al, J. Am. Chem. Soc. 2006 Feb 8; 128(5): 1705-10, herein incorporated by reference).
- the theory behind nanopore sequencing has to do with what occurs when a nanopore is immersed in a conducting fluid and a potential (voltage) is applied across it. Under these conditions a slight electric current due to conduction of ions through the nanopore can be observed, and the amount of current is exceedingly sensitive to the size of the nanopore.
- As each base of a nucleic acid passes through the nanopore this causes a change in the magnitude of the current through the nanopore that is distinct for each of the four bases, thereby allowing the sequence of the DNA molecule to be determined.
- the technology finds use in HeliScope by Helicos Biosciences (Voelkerding et al., Clinical Chem., 55: 641-658, 2009; MacLean et al., Nature Rev. Microbiol., 7: 287-296; U.S. Pat. No. 7,169,560; U.S. Pat. No. 7,282,337; U.S. Pat. No. 7,482,120; U.S. Pat. No. 7,501,245; U.S. Pat. No. 6,818,395; U.S. Pat. No. 6,911,345; U.S. Pat. No. 7,501,245; each herein incorporated by reference in their entirety).
- Template DNA is fragmented and polyadenylated at the 3' end, with the final adenosine bearing a fluorescent label.
- Denatured polyadenylated template fragments are ligated to poly(dT) oligonucleotides on the surface of a flow cell.
- Initial physical locations of captured template molecules are recorded by a CCD camera, and then label is cleaved and washed away.
- Sequencing is achieved by addition of polymerase and serial addition of fluorescently -labeled dNTP reagents. Incorporation events result in fluor signal corresponding to the dNTP, and signal is captured by a CCD camera before each round of dNTP addition.
- Sequence read length ranges from 25-50 nucleotides, with overall output exceeding 1 billion nucleotide pairs per analytical run.
- the Ion Torrent technology is a method of DNA sequencing based on the detection of hydrogen ions that are released during the polymerization of DNA (see, e.g., Science 327(5970): 1190 (2010); U.S. Pat. Appl. Pub. Nos. 20090026082, 20090127589, 20100301398, 20100197507, 20100188073, and 20100137143, incorporated by reference in their entireties for all purposes).
- a microwell contains a template DNA strand to be sequenced. Beneath the layer of microwells is a hypersensitive ISFET ion sensor. All layers are contained within a CMOS semiconductor chip, similar to that used in the electronics industry.
- a hydrogen ion is released, which triggers a hypersensitive ion sensor.
- a hydrogen ion is released, which triggers a hypersensitive ion sensor.
- multiple dNTP molecules will be incorporated in a single cycle. This leads to a corresponding number of released hydrogens and a proportionally higher electronic signal.
- This technology differs from other sequencing technologies in that no modified nucleotides or optics are used.
- the per-base accuracy of the Ion Torrent sequencer is -99.6% for 50 base reads, with -100 Mb to 100Gb generated per run.
- the read-length is 100-300 base pairs.
- the accuracy for homopolymer repeats of 5 repeats in length is -98%.
- the benefits of ion semiconductor sequencing are rapid sequencing speed and low upfront and operating costs.
- the technology finds use in another nucleic acid sequencing approach developed by Stratos Genomics, Inc. and involves the use of Xpandomers.
- This sequencing process typically includes providing a daughter strand produced by a template-directed synthesis.
- the daughter strand generally includes a plurality of subunits coupled in a sequence corresponding to a contiguous nucleotide sequence of all or a portion of a target nucleic acid in which the individual subunits comprise a tether, at least one probe or nucleobase residue, and at least one selectively cleavable bond.
- the selectively cleavable bond(s) is/are cleaved to yield an Xpandomer of a length longer than the plurality of the subunits of the daughter strand.
- the Xpandomer typically includes the tethers and reporter elements for parsing genetic information in a sequence corresponding to the contiguous nucleotide sequence of all or a portion of the target nucleic acid. Reporter elements of the
- Xpandomer are then detected. Additional details relating to Xpandomer-based approaches are described in, for example, U.S. Pat. Pub No. 20090035777, entitled “High Throughput Nucleic Acid Sequencing by Expansion,” filed June 19, 2008, which is incorporated herein in its entirety.
- nucleic acid hybridization techniques include, but are not limited to, in situ hybridization (ISH), microarray, and Southern or Northern blot.
- ISH can also use two or more probes, labeled with radioactivity or the other non-radioactive labels, to simultaneously detect two or more transcripts.
- the present disclosure further provides a method of performing a FISH assay on the patient sample.
- the methods disclosed herein may comprise performing a FISH assay on one or more cells, tissues, organs, or fluids surrounding such cells, tissues and organs.
- the methods disclosed herein further comprise performing a FISH assy on human prostate cells, human prostate tissue or on the fluid surrounding said human prostate cells or human prostate tissue.
- the methods disclosed herien comprise performing a FISH assay on breast cells, lung cells, pancreatic cells, liver cells, breast tissue, lung tissue, pancreatic tissue, liver tissue, or on the fluid surrounding the cells or tissues. Specific protocols are well known in the art and can be readily adapted for the present disclosure.
- kits that are commercially available and that provide protocols for performing FISH assays (available from e.g. , Oncor, Inc., Gaithersburg, MD).
- Patents providing guidance on methodology include U.S. 5,225,326; 5,545,524; 6,121,489 and 6,573,043. All of these references are hereby incorporated by reference in their entirety and may be used along with similar references in the art and with the information provided in the Examples section herein to establish procedural steps convenient for a particular laboratory.
- the one or more ncRNAs may be detected by conducting one or more hybridization reactions.
- the one or more hybridization reactions may comprise one or more hybridization arrays, hybridization reactions, hybridization chain reactions, isothermal hybridization reactions, nucleic acid hybridization reactions, or a combination thereof.
- the one or more hybridization arrays may comprise hybridization array genotyping, hybridization array proportional sensing, DNA hybridization arrays, macroarrays, microarrays, high-density oligonucleotide arrays, genomic hybridization arrays, comparative hybridization arrays, or a combination thereof.
- microarrays including, but not limited to:
- Microarrays can be used to identify disease genes or transcripts (e.g., ncRNAs) by comparing gene expression in disease and normal cells.
- Microarrays can be fabricated using a variety of technologies, including but not limiting: printing with fine- pointed pins onto glass slides; photolithography using pre-made masks; photolithography using dynamic micromirror devices; ink-jet printing; or, electrochemistry on microelectrode arrays.
- the methods disclosed herein may comprise conducting one or more amplification reactions.
- Nucleic acids e.g., ncRNAs
- Conducting one or more amplification reactions may comprise one or more PCR-based amplifications, non-PCR based amplifications, or a combination thereof.
- TMA Transcription mediated amplification
- a target nucleic acid sequence autocatalytically under conditions of substantially constant temperature, ionic strength, and pH in which multiple RNA copies of the target sequence autocatalytically generate additional copies.
- TMA optionally incorporates the use of blocking moieties, terminating moieties, and other modifying moieties to improve TMA process sensitivity and accuracy.
- the ligase chain reaction (Weiss, R., Science 254: 1292 (1991), herein incorporated by reference in its entirety), commonly referred to as LCR, uses two sets of complementary DNA oligonucleotides that hybridize to adjacent regions of the target nucleic acid.
- LCR ligase chain reaction
- oligonucleotides are covalently linked by a DNA ligase in repeated cycles of thermal denaturation, hybridization and ligation to produce a detectable double-stranded ligated oligonucleotide product.
- Strand displacement amplification (Walker, G. et al, Proc. Natl. Acad. Sci. USA 89: 392-396 (1992); U.S. Pat. Nos. 5,270,184 and 5,455,166, each of which is herein incorporated by reference in its entirety), commonly referred to as SDA, uses cycles of annealing pairs of primer sequences to opposite strands of a target sequence, primer extension in the presence of a dNTPaS to produce a duplex hemiphosphorothioated primer extension product, endonuclease-mediated nicking of a hemimodified restriction endonuclease recognition site, and polymerase-mediated primer extension from the 3' end of the nick to displace an existing strand and produce a strand for the next round of primer annealing, nicking and strand displacement, resulting in geometric amplification of product.
- Thermophilic SDA (tSDA) uses thermophilic endonucleases and polymera
- amplification methods include, for example: nucleic acid sequence based amplification (U.S. Pat. No. 5,130,238, herein incorporated by reference in its entirety), commonly referred to as NASBA; one that uses an RNA replicase to amplify the probe molecule itself (Lizardi et al, BioTechnol. 6: 1197 (1988), herein incorporated by reference in its entirety), commonly referred to as replicase; a transcription based amplification method (Kwoh et al, Proc. Natl. Acad. Sci. USA 86: 1173 (1989)); and, self-sustained sequence replication (Guatelli et al, Proc. Natl. Acad. Sci. USA 87: 1874 (1990), each of which is herein incorporated by reference in its entirety).
- NASBA nucleic acid sequence based amplification
- replicase a transcription based amplification method
- self-sustained sequence replication (Guatelli et al
- a sample e.g., a biopsy or a serum or urine sample
- a profiling service e.g., clinical lab at a medical facility, genomic profiling business, etc.
- the subject may visit a medical center to have the sample obtained and sent to the profiling center, or subjects may collect the sample themselves (e.g.
- the sample comprises previously determined biological information
- the information may be directly sent to the profiling service by the subject (e.g. , an information card containing the information may be scanned by a computer and the data transmitted to a computer of the profiling center using an electronic communication systems).
- the profiling service Once received by the profiling service, the sample is processed and a profile is produced (i.e. , expression data), specific for the diagnostic or prognostic information desired for the subject.
- the profile data is then prepared in a format suitable for interpretation by one or more medical personnel (e.g., a treating clinician, physician assistant, nurse, or pharmacist).
- the prepared format may represent a diagnosis or risk assessment (e.g. , presence or absence of a ncRNA) for the subject, along with recommendations for particular treatment options.
- the data may be displayed to the medical personnel by any suitable method.
- the profiling service generates a report that can be printed for the medical personnel (e.g. , at the point of care) or displayed to the medical personnel on a computer monitor.
- the information is first analyzed at the point of care or at a regional facility.
- the raw data is then sent to a central processing facility for further analysis and/or to convert the raw data to information useful for medical personnel or patient.
- the central processing facility provides the advantage of privacy (all data is stored in a central facility with uniform security protocols), speed, and uniformity of data analysis.
- the central processing facility can then control the fate of the data following treatment of the subject. For example, using an electronic communication system, the central facility can provide data to the medical personnel, the subject, or researchers.
- the subject is able to directly access the data using the electronic communication system.
- the subject may chose further intervention or counseling based on the results.
- compositions for use in the diagnostic methods described herein include, but are not limited to, probes, amplification oligonucleotides, and the like.
- compositions and kits may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more,
- the probes may hybridize to 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more,
- the target molecules may be a ncRNA, RNA, DNA, cDNA, mRNA, a portion or fragment thereof or a combination thereof. In some instances, at least a portion of the target molecules are ncRNAs.
- the probes may hybridize to 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more ncRNAs disclosed herein (e.g., SEQ ID NOs: 1-2309).
- the probes comprise a target specific sequence.
- the target specific sequence may be complementary to at least a portion of the target molecule.
- the target specific sequence may be at least about 50% or more, 55% or more, 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 90% or more, 95% or more, 97% or more, 98% or more, or 100% complementary to at leat a portion of the target molecule.
- the target specific sequence may be at least about 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 16 or more, 17 or more, 18 or more, 19 or more, 20 or more nucleotides in length. In some instances, the target specific sequence is between about 8 to about 20 nucleotides, 10 to about 18 nucleotides, or 12 to about 16 nucleotides in length.
- compositions and kits may comprise a plurality of probes, wherein the two or more probes of the plurality of probes comprise identical target specific sequences.
- compositions and kits may comprise a plurality of probes, wherein the two or more probes of the plurality of probes comprise different target specific sequences.
- the probes may further comprise a unique sequence.
- the unique sequence is
- the unique sequence may comprise a label, barcode, or unique identifier.
- the unique sequence may comprise a random sequence, nonrandom sequence, or a combination thereof.
- the unique sequence may be at least about 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 16 or more, 17 or more, 18 or more, 19 or more, 20 or more, 22 or more, 24 or more, 26 or more, 28 or more, 30 or more nucleotides in length.
- the unique sequence is between about 8 to about 20 nucleotides, 10 to about 18 nucleotides, or 12 to about 16 nucleotides in length.
- the unique sequence may allow differentiation of two or more target molecules.
- the two or more target molecules may have identical sequences.
- the unique sequence may allow quantification of a target molecule.
- the two or more target molecules may have different sequences.
- the unique sequence may allow detection of the target molecules.
- the compositions and kits may comprise a plurality of probes for quantifying one or more target molecules.
- the compositions and kits may comprise a plurality of probes for detecting one or more target molecules.
- the unique sequence may allow differentiation of two or more samples.
- the compositions and kits may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more probe sets for differentiating two or more samples from one or more subjects.
- the two or more samples may be from two or more different subjects.
- the compositions and kits comprise a first set of probes comprising a first unique sequence that is specific for a first subject and a second set of probes comprosing a second unique sequence that is specific for a second subject.
- the compositions and kits may further comprise one or more sets of probes with one or more unique sequences to differentiate one or more additional subjects.
- compositions and kits may comprise 2 or more probe sets for differentiating from 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more samples from 1 or more subjects.
- compositions and kits may comprise 2 or more probe sets for differentiating samples from 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more subjects.
- the two or more samples may be from two or more different timepoints from the same subject or different subjects.
- the compositions and kits comprise a first set of probes comprising a first unique sequence that is specific for a first subject and a second set of probes comprosing a second unique sequence that is specific for a second subject.
- the compositions and kits may comprise 2 or more probe sets for differentiating samples from 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more timepoints.
- the timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more hours.
- the timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more days.
- the timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more weeks.
- the timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more months.
- the timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more years.
- the timepoints may be before diagnosis, after diagnosis, before treatment, during treatment, after treatment, before metastasis, after metastatis, before remission, during remission, or a combination thereof.
- compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are identical and the first unique sequence and the second unique sequence are different.
- the compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are different and the first unique sequence and the second unique sequence are different.
- compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are identical and the first unique sequence and the second unique sequence are identical.
- the compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are different and the first unique sequence and the second unique sequence are identical.
- the probes may further comprise a universal sequence.
- the universal sequence may comprise a primer binding site.
- the universal sequence may enable detection of the target sequence.
- the universal sequence may enable amplification of the target sequence.
- the universal sequence may enable transcription or reverse transcription of the target sequence.
- the universal sequence may enable sequencing of the target sequence.
- the probe and antibody compositions of the present disclosure may also be provided on a solid support.
- the solid support may comprise one or more beads, plates, solid surfaces, wells, chips, or a combination thereof.
- the beads may be magnetic, antibody coated, protein A crosslinked, protein G crosslinked, streptavidin coated, oligonucleotide conjugated, silica coated, or a combination thereof.
- beads include, but are not limited to, Ampure beads, AMPure XP beads, streptavidin beads, agarose beads, magnetic beads, Dynabeads®, MACS® microbeads, antibody conjugated beads (e.g., anti-immunoglobulin microbead), protein A conjugated beads, protein G conjugated beads, protein A/G conjugated beads, protein L conjugated beads, oligo-dT conjugated beads, silica beads, silica-like beads, anti-biotin microbead, anti-fluorochrome microbead, and BcMagTM Carboxy-Terminated Magnetic Beads.
- compositions and kits may comprise primers and primer pairs capable of amplifying target molecules, or fragments or subsequences or complements thereof.
- the nucleotide sequences of the target molecules may be provided in computer-readable media for in silico applications and as a basis for the design of appropriate primers for amplification of one or more target molecules.
- Primers based on the nucleotide sequences of target molecules can be designed for use in amplification of the target molecules. For use in amplification reactions such as PCR, a pair of primers can be used.
- the exact composition of the primer sequences is not critical to the disclosure, but for most applications the primers may hybridize to specific sequences of the target molecules or the universal sequence of the probe under stringent conditions, particularly under conditions of high stringency, as known in the art.
- the pairs of primers are usually chosen so as to generate an amplification product of at least about 15 or more, 20 or more, 30 or more, 40 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 125 or more, 150 or more, 175 or more, 200 or more, 250 or more, 300 or more, 350 or more, 400 or more, 450 or more, 500 or more, 600 or more, 700 or more, 800 or more, 900 or more, or 1000 or more nucleotides.
- primer sequences are generally known, and are available in commercial software packages. These primers may be used in standard quantitative or qualitative PCR-based assays to assess transcript expression levels of target molecules. Alternatively, these primers may be used in combination with probes, such as molecular beacons in amplifications using real-time PCR.
- the primer may comprise nucleotide sequences at the 5' and/or 3' termini that are not derived from the target molecule.
- Nucleotide sequences which are not derived from the nucleotide sequence of the target molecule may provide additional functionality to the primer. For example, they may provide a restriction enzyme recognition sequence or a "tag" that facilitates detection, isolation, purification or immobilization onto a solid support.
- the additional nucleotides may provide a self- complementary sequence that allows the primer to adopt a hairpin configuration. Such configurations may be necessary for certain primers, for example, molecular beacon and Scorpion primers, which can be used in solution hybridization techniques.
- the probes or primers can incorporate moieties useful in detection, isolation, purification, or immobilization, if desired.
- moieties are well-known in the art (see, for example, Ausubel et al, (1997 & updates) Current Protocols in Molecular Biology, Wiley & Sons, New York) and are chosen such that the ability of the probe to hybridize with its target molecule is not affected.
- Suitable moieties are detectable labels, such as radioisotopes, fluorophores, chemiluminophores, enzymes, colloidal particles, and fluorescent microparticles, as well as antigens, antibodies, haptens, avidin/streptavidin, biotin, haptens, enzyme cofactors / substrates, enzymes, and the like.
- a label can optionally be attached to or incorporated into a probe or primer to allow detection and/or quantitation of a target polynucleotide representing the target molecule of interest.
- the target polynucleotide may be the expressed target molecule RNA itself, a cDNA copy thereof, or an amplification product derived therefrom, and may be the positive or negative strand, so long as it can be specifically detected in the assay being used.
- an antibody may be labeled.
- labels used for detecting different target molecules may be distinguishable.
- the label can be attached directly (e.g., via covalent linkage) or indirectly, e.g., via a bridging molecule or series of molecules (e.g., a molecule or complex that can bind to an assay component, or via members of a binding pair that can be incorporated into assay components, e.g. biotin-avidin or streptavidin).
- a bridging molecule or series of molecules e.g., a molecule or complex that can bind to an assay component, or via members of a binding pair that can be incorporated into assay components, e.g. biotin-avidin or streptavidin.
- Many labels are commercially available in activated forms which can readily be used for such conjugation (for example through amine acylation), or labels may be attached through known or determinable conjugation schemes, many of which are known in the art.
- Labels useful in the disclosure described herein include any substance which can be detected when bound to or incorporated into the target molecule. Any effective detection method can be used, including optical, spectroscopic, electrical, piezoelectrical, magnetic, Raman scattering, surface plasmon resonance, colorimetric, calorimetric, etc.
- a label is typically selected from a chromophore, a lumiphore, a fluorophore, one member of a quenching system, a chromogen, a hapten, an antigen, a magnetic particle, a material exhibiting nonlinear optics, a semiconductor nanocrystal, a metal nanoparticle, an enzyme, an antibody or binding portion or equivalent thereof, an aptamer, and one member of a binding pair, and combinations thereof.
- Quenching schemes may be used, wherein a quencher and a fluorophore as members of a quenching pair may be used on a probe, such that a change in optical parameters occurs upon binding to the target introduce or quench the signal from the fluorophore.
- a target polynucleotide may comprise a biotin-binding species, and an optically detectable label may be conjugated to biotin and then bound to the labeled target polynucleotide.
- a polynucleotide sensor may comprise an immunological species such as an antibody or fragment, and a secondary antibody containing an optically detectable label may be added.
- Chromophores useful in the methods described herein include any substance which can absorb energy and emit light.
- a plurality of different signaling chromophores can be used with detectably different emission spectra.
- the chromophore can be a lumophore or a fluorophore.
- Typical fluorophores include fluorescent dyes, semiconductor nanocrystals, lanthanide chelates, polynucleotide-specific dyes and green fluorescent protein.
- Coding schemes may optionally be used, comprising encoded particles and/or encoded tags associated with different polynucleotides of the disclosure.
- a variety of different coding schemes are known in the art, including fluorophores, including SCNCs, deposited metals, and RF tags.
- Polynucleotides from the described target molecules may be employed as probes for detecting target molecules expression, for ligation amplification schemes, or may be used as primers for amplification schemes of all or a portion of a target molecule. When amplified, either strand produced by amplification may be provided in purified and/or isolated form.
- compositions and kits comprise a biomarker library.
- the biomarker library may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more target molecules.
- the target molecules may be a ncRNA, RNA, DNA, cDNA, mRNA, a portion or fragment thereof or a combination thereof.
- the biomarker library may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more ncRNAs disclosed herein.
- a kit for analyzing a cancer comprising (a) a probe set comprising a plurality of probes comprising target specific sequences complementary to one or more target molecules, wherein the one or more target molecules comprise one or more ncRNAs; and (b) a computer model or algorithm for analyzing an expression level and/or expression profile of the one or more target molecules in a sample.
- the target molecules may comprose one or more of those described by SEQ ID NOs: 1 -2309, or a combination thereof.
- a kit for analyzing a cancer comprising (a) a probe set comprising a plurality of probes comprising target specific sequences complementary to one or more target molecules of a biomarker library; and (b) a computer model or algorithm for analyzing an expression level and/or expression profile of the one or more target molecules in a sample.
- Control samples and/or nucleic acids may optionally be provided in the kit.
- Control samples may include tissue and/or nucleic acids obtained from or representative of tumor samples from a healthy subject, as well as tissue and/or nucleic acids obtained from or representative of tumor samples from subjects diagnosed with a cancer.
- the devices may include an excitation and/or a detection means. Any instrument that provides a wavelength that can excite a species of interest and is shorter than the emission wavelength(s) to be detected can be used for excitation. Commercially available devices can provide suitable excitation wavelengths as well as suitable detection component.
- Exemplary excitation sources include a broadband UV light source such as a deuterium lamp with an appropriate filter, the output of a white light source such as a xenon lamp or a deuterium lamp after passing through a monochromator to extract out the desired wavelength(s), a continuous wave (cw) gas laser, a solid state diode laser, or any of the pulsed lasers.
- Emitted light can be detected through any suitable device or technique; many suitable approaches are known in the art.
- a fluorimeter or spectrophotometer may be used to detect whether the test sample emits light of a wavelength characteristic of a label used in an assay.
- the devices typically comprise a means for identifying a given sample, and of linking the results obtained to that sample.
- Such means can include manual labels, barcodes, and other indicators which can be linked to a sample vessel, and/or may optionally be included in the sample itself, for example where an encoded particle is added to the sample.
- the results may be linked to the sample, for example in a computer memory that contains a sample designation and a record of expression levels obtained from the sample. Linkage of the results to the sample can also include a linkage to a particular sample receptacle in the device, which is also linked to the sample identity.
- the device also comprises output means for outputting the disease status, prognosis and/or a treatment modality.
- output means can take any form which transmits the results to a patient and/or a healthcare provider, and may include a monitor, a printed format, or both.
- the device may use a computer system for performing one or more of the steps provided.
- the methods disclosed herein may also comprise the transmission of data/information.
- data/information derived from the detection and/or quantification of the target may be transmitted to another device and/or instrument.
- the information obtained from an algorithm may also be transmitted to another device and/or instrument.
- Transmission of the data/information may comprise the transfer of data/information from a first source to a second source.
- the first and second sources may be in the same approximate location (e.g., within the same room, building, block, campus). Alternatively, first and second sources may be in multiple locations (e.g., multiple cities, states, countries, continents, etc).
- Transmission of the data/information may comprise digital transmission or analog transmission.
- Digital transmission may comprise the physical transfer of data (a digital bit stream) over a point-to-point or point-to-multipoint communication channel. Examples of such channels are copper wires, optical fibres, wireless communication channels, and storage media.
- the data may be represented as an electromagnetic signal, such as an electrical voltage, radiowave, microwave, or infrared signal.
- Analog transmission may comprise the transfer of a continuously varying analog signal.
- the messages can either be represented by a sequence of pulses by means of a line code (baseband transmission), or by a limited set of continuously varying wave forms (passband transmission), using a digital modulation method.
- the passband modulation and corresponding demodulation also known as detection
- modem equipment According to the most common definition of digital signal, both baseband and passband signals representing bit-streams are considered as digital transmission, while an alternative definition only considers the baseband signal as digital, and passband transmission of digital data as a form of digital-to-analog conversion,
- Samples for use with the compositions and kits and in the methods of the present disclosure comprise nucleic acids suitable for providing RNA expression information.
- the biological sample from which the expressed RNA is obtained and analyzed for target molecule expression can be any material suspected of comprising cancer tissue or cells.
- the sample can be a biological sample used directly in a method of the disclosure.
- the sample can be a sample prepared from a biological sample.
- the sample or portion of the sample comprising or suspected of comprising cancer tissue or cells can be any source of biological material, including cells, tissue, secretions, or fluid, including bodily fluids.
- the source of the sample include an aspirate, a needle biopsy, a cytology pellet, a bulk tissue preparation or a section thereof obtained for example by surgery or autopsy, lymph fluid, blood, plasma, serum, tumors, and organs.
- the source of the sample can be urine, bile, excrement, sweat, tears, vaginal fluids, spinal fluid, and stool.
- the sources of the sample are secretions.
- the secretions are exosomes.
- the samples may be archival samples, having a known and documented medical outcome, or may be samples from current patients whose ultimate medical outcome is not yet known.
- the sample may be dissected prior to molecular analysis.
- the sample may be prepared via macrodissection of a bulk tumor specimen or portion thereof, or may be treated via microdissection, for example via Laser Capture Microdissection (LCM).
- LCD Laser Capture Microdissection
- the sample may initially be provided in a variety of states, as fresh tissue, fresh frozen tissue, fine needle aspirates, and may be fixed or unfixed. Frequently, medical laboratories routinely prepare medical samples in a fixed state, which facilitates tissue storage.
- fixatives can be used to fix tissue to stabilize the morphology of cells, and may be used alone or in combination with other agents. Exemplary fixatives include crosslinking agents, alcohols, acetone, Bouin's solution, Zenker solution, Hely solution, osmic acid solution and Camoy solution.
- Crosslinking fixatives can comprise any agent suitable for forming two or more covalent bonds, for example, an aldehyde.
- Sources of aldehydes typically used for fixation include formaldehyde, paraformaldehyde, glutaraldehyde or formalin.
- the crosslinking agent comprises formaldehyde, which may be included in its native form or in the form of
- One or more alcohols may be used to fix tissue, alone or in combination with other fixatives.
- exemplary alcohols used for fixation include methanol, ethanol and isopropanol.
- Formalin fixation is frequently used in medical laboratories.
- Formalin comprises both an alcohol, typically methanol, and formaldehyde, both of which can act to fix a biological sample.
- the biological sample may optionally be embedded in an embedding medium.
- embedding media used in histology including paraffin, Tissue-Tek® V.I.P.TM, Paramat, Paramat Extra, Paraplast, Paraplast X-tra, Paraplast Plus, Peel Away Paraffin Embedding Wax, Polyester Wax, Carbowax Polyethylene Glycol, PolyfinTM, Tissue Freezing Medium TFMFM, Cryo-GefTM, and OCT Compound (Electron Microscopy Sciences, Hatfield, PA).
- the embedding material may be removed via any suitable techniques, as known in the art.
- the sample is a fixed, wax-embedded biological sample.
- samples from medical laboratories are provided as fixed, wax-embedded samples, most commonly as formalin-fixed, paraffin embedded (FFPE) tissues.
- FFPE formalin-fixed, paraffin embedded
- the target polynucleotide that is ultimately assayed can be prepared synthetically (in the case of control sequences), but typically is purified from the biological source and subjected to one or more preparative steps.
- the RNA may be purified to remove or diminish one or more undesired components from the biological sample or to concentrate it. Conversely, where the RNA is too concentrated for the particular assay, it may be diluted.
- the present disclosure provides drug screening assays (e.g. , to screen for anticancer drugs).
- the screening methods of the present disclosure utilize ncRNAs.
- the present disclosure provides methods of screening for compounds that alter the expression or activity of ncRNAs.
- the compounds may increase the expression or activity of the ncRNAs.
- the compounds may decrease the expression or activity of the ncRNAs.
- the compounds or agents may interfere with transcription, by interacting, for example, with the promoter region.
- the compounds or agents may interfere with mRNA (e.g. , by RNA interference, antisense technologies, etc.).
- the compounds or agents may interfere with pathways that are upstream or downstream of the biological activity of ncRNAs.
- candidate compounds are antisense or interfering RNA agents (e.g. , oligonucleotides) directed against ncRNAs.
- candidate compounds are antibodies or small molecules that specifically bind to a ncRNA regulator.
- the candidate compounds are expression products that inhibit thebiological function of the ncRNAs.
- candidate compounds are evaluated for their ability to alter ncRNAs expression by contacting a compound with a cell expressing a ncRNA and then assaying for the effect of the candidate compounds on expression.
- the effect of candidate compounds on expression of ncRNAs is assayed for by detecting the level ncRNA expressed by the cell.
- mRNA expression can be detected by any suitable method.
- the methods, compositions, and kits disclosed herein may be used for the diagnosis, prognosis, and/or monitoring the status or outcome of a cancer in a subject.
- the diagnosing, predicting, and/or monitoring the status or outcome of a cancer comprises determining the malignancy or malignant potential of the cancer or tumor.
- the diagnosing, predicting, and/or monitoring the status or outcome of a cancer comprises determining the stage of the cancer.
- the diagnosing, predicting, and/or monitoring the status or outcome of a cancer can comprise determining the tumor grade.
- the diagnosing, predicting, and/or monitoring the status or outcome of a cancer comprises assessing the risk of developing a cancer.
- the diagnosing, predicting, and/or monitoring the status or outcome of a cancer includes assessing the risk of cancer recurrence. In some embodiments, diagnosing, predicting, and/or monitoring the status or outcome of a cancer may comprise determining the efficacy of treatment.
- diagnosing, predicting, and/or monitoring the status or outcome of a cancer may comprise determining a therapeutic regimen. Determining a therapeutic regimen may comprise administering an anti-cancer therapeutic. Alternatively, determining the treatment for the cancer may comprise modifying a therapeutic regimen. Modifying a therapeutic regimen may comprise increasing, decreasing, or terminating a therapeutic regimen.
- the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 50%. In other instances, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 60%. The methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 65%. Alternatively, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 70%.
- the disclosure also encompasses any of the methods disclosed herein where the sensitivity is at least about 45%. In some embodiments, the sensitivity is at least about 50%. In some embodiments, the sensitivity is at least about 55%. In some embodiments, the sensitivity is at least about 60%. In some embodiments, the sensitivity is at least about 65%. In some embodiments, the sensitivity is at least about 70%. In some embodiments, the sensitivity is at least about 75%. In some embodiments, the sensitivity is at least about 80%. In some embodiments, the sensitivity is at least about 85%. In some embodiments, the sensitivity is at least about 90%. In some embodiments, the sensitivity is at least about 95%.
- the disclosure also encompasses any of the methods disclosed herein where the expression level determines the status or outcome of a cancer in the subject with at least about 45% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 50% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 55% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 60% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 65% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 70% specificity.
- the expression level determines the status or outcome of a cancer in the subject with at least about 75% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 80% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 85% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 90% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 95% specificity.
- a cancer is characterized by the uncontrolled growth of abnormal cells anywhere in a body.
- the abnormal cells may be termed cancer cells, malignant cells, or tumor cells.
- Many cancers and the abnormal cells that compose the cancer tissue are further identified by the name of the tissue that the abnormal cells originated from (for example, breast cancer, lung cancer, colon cancer, prostate cancer, pancreatic cancer, thyroid cancer). Cancer is not confined to humans; animals and other living organisms can get cancer.
- the cancer may be malignant.
- the cancer may be benign.
- the cancer may be a recurrent and/or refractory cancer. Most cancers can be classified as a carcinoma, sarcoma, leukemia, lymphoma, myeloma, or a central nervous system cancer.
- the cancer may be a sarcoma.
- Sarcomas are cancers of the bone, cartilage, fat, muscle, blood vessels, or other connective or supportive tissue.
- Sarcomas include, but are not limited to, bone cancer, fibrosarcoma, chondrosarcoma, Ewing's sarcoma, malignant hemangioendothelioma, malignant schwannoma, bilateral vestibular schwannoma, osteosarcoma, soft tissue sarcomas (e.g.
- alveolar soft part sarcoma alveolar soft part sarcoma, angiosarcoma, cystosarcoma phylloides, dermatofibrosarcoma, desmoid tumor, epithelioid sarcoma, extraskeletal osteosarcoma, fibrosarcoma, hemangiopericytoma, hemangiosarcoma, Kaposi's sarcoma, leiomyosarcoma, liposarcoma, lymphangiosarcoma, lymphosarcoma, malignant fibrous histiocytoma, neurofibrosarcoma, rhabdomyosarcoma, and synovial sarcoma).
- the cancer may be a carcinoma.
- Carcinomas are cancers that begin in the epithelial cells, which are cells that cover the surface of the body, produce hormones, and make up glands.
- carcinomas include breast cancer, pancreatic cancer, lung cancer, colon cancer, colorectal cancer, rectal cancer, kidney cancer, bladder cancer, stomach cancer, prostate cancer, liver cancer, ovarian cancer, brain cancer, vaginal cancer, vulvar cancer, uterine cancer, oral cancer, penic cancer, testicular cancer, esophageal cancer, skin cancer, cancer of the fallopian tubes, head and neck cancer, gastrointestinal stromal cancer, adenocarcinoma, cutaneous or intraocular melanoma, cancer of the anal region, cancer of the small intestine, cancer of the endocrine system, cancer of the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal gland, cancer of the urethra, cancer of the renal pelvis, cancer of the ureter, cancer of the
- the cancer is a skin cancer, such as a basal cell carcinoma, squamous, melanoma, nonmelanoma, or actinic (solar) keratosis.
- the cancer is a prostate cancer.
- the cancer may be a thyroid cancer.
- the cancer can be a pancreatic cancer.
- the cancer is a bladder cancer.
- the cancer is a lung cancer.
- Lung cancer can start in the airways that branch off the trachea to supply the lungs (bronchi) or the small air sacs of the lung (the alveoli).
- Lung cancers include non-small cell lung carcinoma (NSCLC), small cell lung carcinoma, and mesotheliomia.
- NSCLC non-small cell lung carcinoma
- Examples of NSCLC include squamous cell carcinoma, adenocarcinoma, and large cell carcinoma.
- the mesothelioma may be a cancerous tumor of the lining of the lung and chest cavity (pleura) or lining of the abdomen (peritoneum). The mesothelioma may be due to asbestos exposure.
- the cancer may be a brain cancer, such as a glioblastoma.
- the cancer may be a central nervous system (CNS) tumor.
- CNS tumors may be classified as gliomas or nongliomas.
- the glioma may be malignant glioma, high grade glioma, diffuse intrinsic pontine glioma. Examples of gliomas include astrocytomas, oligodendrogliomas (or mixtures of oligodendroglioma and astocytoma elements), and ependymomas.
- Astrocytomas include, but are not limited to, low-grade astrocytomas, anaplastic astrocytomas, glioblastoma multiforme, pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and subependymal giant cell astrocytoma.
- Oligodendrogliomas include low-grade oligodendrogliomas (or oligoastrocytomas) and anaplastic oligodendriogliomas.
- Nongliomas include meningiomas, pituitary adenomas, primary CNS lymphomas, and medulloblastomas. In some instances, the cancer is a meningioma.
- the cancer may be leukemia.
- the leukemia may be an acute lymphocytic leukemia, acute myelocytic leukemia, chronic lymphocytic leukemia, or chronic myelocytic leukemia. Additional types of leukemias include hairy cell leukemia, chronic myelomonocytic leukemia, and juvenile myelomonocytic-leukemia.
- the cancer is a lymphoma.
- Lymphomas are cancers of the lymphocytes and may develop from either B or T lymphocytes.
- the two major types of lymphoma are Hodgkin's lymphoma, previously known as Hodgkin's disease, and non-Hodgkin's lymphoma.
- Hodgkin's lymphoma is marked by the presence of the Reed-Sternberg cell.
- Non-Hodgkin's lymphomas are all lymphomas which are not Hodgkin's lymphoma.
- Non-Hodgkin lymphomas may be indolent lymphomas and aggressive lymphomas.
- Non-Hodgkin's lymphomas include, but are not limited to, diffuse large B cell lymphoma, follicular lymphoma, mucosa-associated lymphatic tissue lymphoma (MALT), small cell lymphocytic lymphoma, mantle cell lymphoma, Burkitt's lymphoma, mediastinal large B cell lymphoma, Waldenstrom macroglobulinemia, nodal marginal zone B cell lymphoma (NMZL), splenic marginal zone lymphoma (SMZL), extranodal marginal zone B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, and lymphomatoid granulomatosis.
- MALT mucosa-associated lymphatic tissue lymphoma
- MALT mucosa-associated lymphatic tissue lymphoma
- small cell lymphocytic lymphoma mantle cell lymphoma
- Burkitt's lymphoma mediastinal large B cell
- Diagnosing, predicting, or monitoring a status or outcome of a cancer may comprise determining the stage of the cancer.
- the stage of a cancer is a description (usually numbers I to IV with IV having more progression) of the extent the cancer has spread.
- the stage often takes into account the size of a tumor, how deeply it has penetrated, whether it has invaded adjacent organs, how many lymph nodes it has metastasized to (if any), and whether it has spread to distant organs.
- Staging of cancer can be used as a predictor of survival, and cancer treatment may be determined by staging. Determining the stage of the cancer may occur before, during, or after treatment. The stage of the cancer may also be determined at the time of diagnosis.
- Cancer staging can be divided into a clinical stage and a pathologic stage.
- Cancer staging may comprise the TNM classification.
- TNM Classification of Malignant Tumours is a cancer staging system that describes the extent of cancer in a patient's body. T may describe the size of the tumor and whether it has invaded nearby tissue, N may describe regional lymph nodes that are involved, and M may describe distant metastasis (spread of cancer from one body part to another).
- TNM Tumor, Node, Metastasis
- clinical stage and pathologic stage are denoted by a small "c" or "p" before the stage (e.g., CT3N1M0 or pT2N0).
- Clinical stage may be based on all of the available information obtained before a surgery to remove the tumor. Thus, it may include information about the tumor obtained by physical examination, radiologic examination, and endoscopy.
- Pathologic stage can add additional information gained by examination of the tumor microscopically by a pathologist.
- Pathologic staging can allow direct examination of the tumor and its spread, contrasted with clinical staging which may be limited by the fact that the information is obtained by making indirect observations at a tumor which is still in the body.
- the TNM staging system can be used for most forms of cancer.
- staging may comprise Ann Arbor staging.
- Ann Arbor staging is the staging system for lymphomas, both in Hodgkin's lymphoma (previously called Hodgkin's disease) and Non-Hodgkin lymphoma (abbreviated NHL).
- the stage may depend on both the place where the malignant tissue is located (as located with biopsy, CT scanning and increasingly positron emission tomography) and on systemic symptoms due to the lymphoma ("B symptoms": night sweats, weight loss of >10% or fevers).
- B symptoms night sweats, weight loss of >10% or fevers
- the principal stage may be determined by location of the tumor.
- Stage I may indicate that the cancer is located in a single region, usually one lymph node and the surrounding area. Stage I often may not have outward symptoms.
- Stage II can indicate that the cancer is located in two separate regions, an affected lymph node or organ and a second affected area, and that both affected areas are confined to one side of the diaphragm - that is, both are above the diaphragm, or both are below the diaphragm.
- Stage III often indicates that the cancer has spread to both sides of the diaphragm, including one organ or area near the lymph nodes or the spleen.
- Stage IV may indicate diffuse or disseminated involvement of one or more extralymphatic organs, including any involvement of the liver, bone marrow, or nodular involvement of the lungs.
- Modifiers may also be appended to some stages.
- a or B may indicate the absence of constitutional (B-type) symptoms is denoted by adding an "A” to the stage; the presence is denoted by adding a "B” to the stage.
- E can be used if the disease is "extranodal” (not in the lymph nodes) or has spread from lymph nodes to adjacent tissue.
- X is often used if the largest deposit is >10 cm large (“bulky disease”), or whether the mediastinum is wider than 1/3 of the chest on a chest X-ray.
- S may be used if the disease has spread to the spleen.
- CS may denote that the clinical stage as obtained by doctor's examinations and tests.
- PS may denote that the pathological stage as obtained by exploratory laparotomy (surgery performed through an abdominal incision) with splenectomy (surgical removal of the spleen).
- Diagnosing, predicting, or monitoring a status or outcome of a cancer may comprise treating a cancer or preventing a cancer progression.
- diagnosing, predicting, or monitoring a status or outcome of a cancer may comprise identifying or predicting responders to an anti-cancer therapy.
- diagnosing, predicting, or monitoring may comprise determining a therapeutic regimen. Determining a therapeutic regimen may comprise administering an anti-cancer therapy. Alternatively, determining a therapeutic regimen may comprise modifying, recommending, continuing or discontinuing an anti-cancer regimen.
- the expression patterns can be used to designate one or more treatment modalities (e.g., therapeutic regimens, anti-cancer regimen).
- An anti-cancer regimen may comprise one or more anti-cancer therapies. Examples of anti-cancer therapies include targeting cancer therapy (e.g., targeting the non- coding RNAs described herein), surgery, chemotherapy, radiation therapy,
- the present disclsoure targets the expression of cancer markers.
- the present disclsoure employs compositions comprising oligomeric antisense or RNAi compounds, particularly oligonucleotides (e.g. , those identified in the drug screening methods described above), for use in modulating the function of nucleic acid molecules encoding cancer markers of the present disclsoure, ultimately modulating the amount of cancer marker expressed.
- RNAi is utilized to target non-coding RNAs (e.g., one or more of SEQ
- RNA-induced silencing complex RNA-induced silencing complex
- siRNAs Chemically synthesized siRNAs have become powerful reagents for genome-wide analysis of mammalian gene function in cultured somatic cells. Beyond their value for validation of gene function, siRNAs also hold great potential as gene-specific therapeutic agents (Tuschl and Borkhardt, Molecular Intervent. 2002; 2(3): 158-67, herein incorporated by reference).
- siRNAs are extraordinarily effective at lowering the amounts of targeted RNA, and by extension proteins, frequently to undetectable levels.
- the silencing effect can last several months, and is extraordinarily specific, because one nucleotide mismatch between the target RNA and the central region of the siRNA is frequently sufficient to prevent silencing (Brummelkamp et al, Science 2002; 296:550-3; and Holen et al, Nucleic Acids Res. 2002; 30: 1757-66, both of which are herein incorporated by reference).
- siRNAs An important factor in the design of siRNAs is the presence of accessible sites for siRNA binding.
- Bahoia et al. (J. Biol. Chem, 2003; 278: 15991-15997; herein incorporated by reference) describe the use of a type of DNA array called a scanning array to find accessible sites in mRNAs for designing effective siRNAs.
- These arrays comprise oligonucleotides ranging in size from monomers to a certain maximum, usually Comers, synthesized using a physical barrier (mask) by stepwise addition of each base in the sequence. Thus the arrays represent a full oligonucleotide complement of a region of the target gene.
- Hybridization of the target mRNA to these arrays provides an exhaustive accessibility profile of this region of the target mRNA.
- Such data are useful in the design of antisense oligonucleotides (ranging from 7mers to 25mers), where it is important to achieve a compromise between oligonucleotide length and binding affinity, to retain efficacy and target specificity (Sohail et al, Nucleic Acids Res., 2001; 29(10): 2041- 2045). Additional methods and concerns for selecting siRNAs are described for example, in WO 05054270, WO05038054A1, WO03070966A2, J Mol Biol. 2005 May 13;348(4):883-93, J Mol Biol.
- expression of non-coding RNAs is modulated using antisense compounds that specifically hybridize with one or more nucleic acids encoding the RNAs.
- antisense compounds that specifically hybridize with one or more nucleic acids encoding the RNAs.
- the specific hybridization of an oligomeric compound with its target nucleic acid interferes with the normal function of the nucleic acid. This modulation of function of a target nucleic acid by compounds that specifically hybridize to it is generally referred to as
- RNA to be interfered with include all vital functions such as, for example, translocation of the RNA to the site of protein translation, translation of protein from the RNA, splicing of the RNA to yield one or more mRNA species, and catalytic activity that may be engaged in or facilitated by the RNA.
- the overall effect of such interference with target nucleic acid function is modulation of the expression of cancer markers of the present disclsoure.
- modulation means either an increase (stimulation) or a decrease (inhibition) in the expression of a gene. For example, expression may be inhibited to potentially prevent tumor proliferation.
- Targeting an antisense compound to a particular nucleic acid is a multistep process. The process usually begins with the identification of a nucleic acid sequence whose function is to be modulated. This may be, for example, a cellular gene (or mRNA transcribed from the gene) whose expression is associated with a particular disorder or disease state, or a nucleic acid molecule from an infectious agent.
- the target is a nucleic acid molecule encoding a cancer marker of the present disclsoure.
- the targeting process also includes determination of a site or sites within this gene for the antisense interaction to occur such that the desired effect, e.g., detection or modulation of expression of the protein, will result.
- a preferred intragenic site is the region encompassing the translation initiation or termination codon of the open reading frame (ORF) of the gene. Since the translation initiation codon is typically 5'-AUG (in transcribed mRNA molecules; 5'-ATG in the corresponding DNA molecule), the translation initiation codon is also referred to as the "AUG codon,” the “start codon” or the "AUG start codon”.
- translation initiation codon having the RNA sequence 5'-GUG, 5'-UUG or 5'-CUG, and 5'-AUA, 5'-ACG and 5'-CUG have been shown to function in vivo.
- the terms "translation initiation codon” and "start codon” can encompass many codon sequences, even though the initiator amino acid in each instance is typically methionine (in eukaryotes) or formylmethionine (in prokaryotes).
- Eukaryotic and prokaryotic genes may have two or more alternative start codons, any one of which may be preferentially utilized for translation initiation in a particular cell type or tissue, or under a particular set of conditions.
- start codon and “translation initiation codon” refer to the codon or codons that are used in vivo to initiate translation of an RNA (e.g., one or more of SEQ ID NOs: 1-2309).
- Translation termination codon (or "stop codon") of a gene may have one of three sequences (/. e. , 5'-UAA, 5'-UAG and 5'-UGA; the corresponding DNA sequences are 5'-TAA, 5'-TAG and
- start codon region and “translation initiation codon region” refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction (i.e. , 5' or 3') from a translation initiation codon.
- stop codon region and “translation termination codon region” refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction (i.e. , 5' or 3') from a translation termination codon.
- Other target regions include the 5' untranslated region (5' UTR), referring to the portion of an mRNA in the 5' direction from the translation initiation codon, and thus including nucleotides between the 5' cap site and the translation initiation codon of an mRNA or corresponding nucleotides on the gene, and the 3' untranslated region (3' UTR), referring to the portion of an mRNA in the 3' direction from the translation termination codon, and thus including nucleotides between the translation termination codon and 3' end of an mRNA or corresponding nucleotides on the gene.
- 5' UTR 5' untranslated region
- 3' UTR 3' untranslated region
- the 5' cap of an mRNA comprises an N7-methylated guanosine residue joined to the 5'-most residue of the mRNA via a 5'-5' triphosphate linkage.
- the 5' cap region of an mRNA is considered to include the 5' cap structure itself as well as the first 50 nucleotides adjacent to the cap.
- the cap region may also be a preferred target region.
- mRNA splice sites i.e. , intron-exon junctions
- introns can also be effective, and therefore preferred, target regions for antisense compounds targeted, for example, to DNA or pre-mRNA.
- target sites for antisense inhibition are identified using commercially available software programs (e.g., Biognostik, Gottingen, Germany; SysArris Software, Bangalore, India; Antisense Research Group, University of Liverpool, Liverpool, England; GeneTrove,
- target sites for antisense inhibition are identified using the accessible site method described in PCT Publ. No. WO0198537A2, herein incorporated by reference.
- oligonucleotides are chosen that are sufficiently complementary to the target (i.e. , hybridize sufficiently well and with sufficient specificity) to give the desired effect.
- antisense oligonucleotides are targeted to or near the start codon.
- hybridization means hydrogen bonding, which may be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between complementary nucleoside or nucleotide bases.
- adenine and thymine are complementary nucleobases that pair through the formation of hydrogen bonds. It is understood that the sequence of an antisense compound need not be 100% complementary to that of its target nucleic acid to be specifically hybridizable.
- An antisense compound is specifically hybridizable when binding of the compound to the target DNA or RNA molecule interferes with the normal function of the target DNA or RNA to cause a loss of utility, and there is a sufficient degree of complementarity to avoid non-specific binding of the antisense compound to non-target sequences under conditions in which specific binding is desired (i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and in the case of in vitro assays, under conditions in which the assays are performed).
- antisense oligonucleotides have been employed as therapeutic moieties in the treatment of disease states in animals and man. Antisense oligonucleotides have been safely and effectively administered to humans and numerous clinical trials are presently underway. It is thus established that oligonucleotides are useful therapeutic modalities that can be configured to be useful in treatment regimes for treatment of cells, tissues, and animals, especially humans.
- antisense oligonucleotides are a preferred form of antisense compound
- the present disclsoure comprehends other oligomeric antisense compounds, including but not limited to oligonucleotide mimetics such as are described below.
- the antisense compounds in accordance with this disclsoure preferably comprise from about 8 to about 30 nucleobases (i.e., from about 8 to about 30 linked bases), although both longer and shorter sequences may find use with the present disclsoure.
- Particularly preferred antisense compounds are antisense oligonucleotides, even more preferably those comprising from about 12 to about 25 nucleobases.
- oligonucleotides containing modified backbones or non-natural intemucleoside linkages include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone.
- modified oligonucleotides that do not have a phosphorus atom in their intemucleoside backbone can also be considered to be oligonucleosides.
- Preferred modified oligonucleotide backbones include, for example, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotriesters, methyl and other alkyl phosphonates including 3'-alkylene phosphonates and chiral phosphonates, phosphinates, phosphoramidates including 3 '-amino phosphoramidate and
- thionoalkylphosphotriesters having normal 3'-5' linkages, 2'-5' linked analogs of these, and those having inverted polarity wherein the adjacent pairs of nucleoside units are linked 3'-5' to 5'-3' or 2'-5' to 5'-2'.
- Various salts, mixed salts and free acid forms are also included.
- the present disclsoure contemplates the use of any genetic manipulation for use in modulating the expression of non-coding RNAs (e.g., one or more of SEQ ID NOs: 1-2309).
- Examples of genetic manipulation include, but are not limited to, gene knockout (e.g., removing the gene encoding the RNA from the chromosome using, for example, recombination), expression of antisense constructs with or without inducible promoters, and the like.
- Delivery of nucleic acid construct to cells in vitro or in vivo may be conducted using any suitable method.
- a suitable method is one that introduces the nucleic acid construct into the cell such that the desired event occurs (e.g., expression of an antisense construct).
- Genetic therapy may also be used to deliver siRNA or other interfering molecules that are expressed in vivo (e.g., upon stimulation by an inducible promoter (e.g., an androgen-responsive promoter)).
- Plasmids carrying genetic information into cells are achieved by any of various methods including, but not limited to, directed injection of naked DNA constructs, bombardment with gold particles loaded with said constructs, and macromolecule mediated gene transfer using, for example, liposomes, biopolymers, and the like.
- Preferred methods use gene delivery vehicles derived from viruses, including, but not limited to, adenoviruses, retroviruses, vaccinia viruses, and adeno-associated viruses. Because of the higher efficiency as compared to retroviruses, vectors derived from adenoviruses are the preferred gene delivery vehicles for transferring nucleic acid molecules into host cells in vivo.
- Adenoviral vectors have been shown to provide very efficient in vivo gene transfer into a variety of solid tumors in animal models and into human solid tumor xenografts in immune-deficient mice. Examples of adenoviral vectors and methods for gene transfer are described in PCT publications WO 00/12738 and WO 00/09675 and U.S. Pat. Appl. Nos. 6,033,908, 6,019,978, 6,001,557, 5,994,132, 5,994,128, 5,994,106, 5,981,225, 5,885,808, 5,872,154, 5,830,730, and 5,824,544, each of which is herein incorporated by reference in its entirety.
- Vectors may be administered to subject in a variety of ways.
- vectors are administered into tumors or tissue associated with tumors using direct injection.
- administration is via the blood or lymphatic circulation (See e.g., PCT publication 99/02685 herein incorporated by reference in its entirety).
- Exemplary dose levels of adenoviral vector are preferably 10 8 to 10 11 vector particles added to the perfusate.
- Surgical oncology uses surgical methods to diagnose, stage, and treat cancer, and to relieve certain cancer-related symptoms.
- Surgery may be used to remove the tumor (e.g., excisions, resections, debulking surgery), reconstruct a part of the body (e.g., restorative surgery), and/or to relieve symptoms such as pain (e.g., palliative surgery).
- Surgery may also include cryosurgery.
- a ball of ice crystals may form around the probe, freezing nearby cells. Sometimes more than one probe is used to deliver the liquid nitrogen to various parts of the tumor.
- the probes may be put into the tumor during surgery or through the skin (percutaneously). After cryosurgery, the frozen tissue thaws and may be naturally absorbed by the body (for internal tumors), or may dissolve and form a scab (for external tumors).
- Chemotherapeutic agents may also be used for the treatment of cancer.
- examples of chemotherapeutic agents include alkylating agents, anti-metabolites, plant alkaloids and terpenoids, vinca alkaloids, podophyllotoxin, taxanes, topoisomerase inhibitors, and cytotoxic antibiotics.
- Cisplatin, carboplatin, and oxaliplatin are examples of alkylating agents.
- Other alkylating agents include mechlorethamine, cyclophosphamide, chlorambucil, ifosfamide.
- Alkylating agens may impair cell function by forming covalent bonds with the amino, carboxyl, sulfhydryl, and phosphate groups in biologically important molecules.
- alkylating agents may chemically modify a cell's DNA.
- Anti-metabolites are another example of chemotherapeutic agents. Anti-metabolites may masquerade as purines or pyrimidines and may prevent purines and pyrimidines from becoming incorporated in to DNA during the "S" phase (of the cell cycle), thereby stopping normal development and division. Antimetabolites may also affect RNA synthesis. Examples of metabolites include azathioprine and mercaptopurine.
- Taxanes may disrupt microtubule function. Microtubules are essential to cell division, and taxanes may stabilize GDP-bound tubulin in the microtubule, thereby inhibiting the process of cell division. Thus, in essence, taxanes may be mitotic inhibitors. Taxanes may also be
- chemotherapeutic agents include podophyllotoxin.
- Podophyllotoxin is a plant- derived compound that may help with digestion and may be used to produce cytostatic drugs such as etoposide and teniposide. They may prevent the cell from entering the Gl phase (the start of DNA replication) and the replication of DNA (the S phase).
- Topoisomerases are essential enzymes that maintain the topology of DNA. Inhibition of type I or type II topoisomerases may interfere with both transcription and replication of DNA by upsetting proper DNA supercoiling. Some chemotherapeutic agents may inhibit topoisomerases.
- some type I topoisomerase inhibitors include camptothecins: irinotecan and topotecan. Examples of type II inhibitors include amsacrine, etoposide, etoposide phosphate, and teniposide.
- chemotherapeutic agents are cytotoxic antibiotics.
- Cytotoxic antibiotics are a group of antibiotics that are used for the treatment of cancer because they may interfere with DNA replication and/or protein synthesis.
- Cytotoxic antiobiotics include, but are not limited to, actinomycin, anthracyclines, doxorubicin, daunorubicin, valrubicin, idarubicin, epirubicin, bleomycin, plicamycin, and mitomycin.
- the anti-cancer treatment may comprise radiation therapy. Radiation can come from a machine outside the body (external-beam radiation therapy) or from radioactive material placed in the body near cancer cells (internal radiation therapy, more commonly called brachytherapy). Systemic radiation therapy uses a radioactive substance, given by mouth or into a vein that travels in the blood to tissues throughout the body.
- External-beam radiation therapy may be delivered in the form of photon beams (either x-rays or gamma rays).
- a photon is the basic unit of light and other forms of electromagnetic radiation.
- An example of external-beam radiation therapy is called 3-dimensional conformal radiation therapy (3D- CRT).
- 3D-CRT may use computer software and advanced treatment machines to deliver radiation to very precisely shaped target areas.
- Many other methods of external -beam radiation therapy are currently being tested and used in cancer treatment. These methods include, but are not limited to, intensity-modulated radiation therapy (IMRT), image-guided radiation therapy (IGRT), Stereotactic radiosurgery (SRS), Stereotactic body radiation therapy (SBRT), and proton therapy.
- IMRT intensity-modulated radiation therapy
- IGRT image-guided radiation therapy
- SRS Stereotactic radiosurgery
- SBRT Stereotactic body radiation therapy
- IMRT Intensity-modulated radiation therapy
- collimators can be stationary or can move during treatment, allowing the intensity of the radiation beams to change during treatment sessions.
- This kind of dose modulation allows different areas of a tumor or nearby tissues to receive different doses of radiation.
- IMRT is planned in reverse (called inverse treatment planning). In inverse treatment planning, the radiation doses to different areas of the tumor and surrounding tissue are planned in advance, and then a high-powered computer program calculates the required number of beams and angles of the radiation treatment.
- IMRT In contrast, during traditional (forward) treatment planning, the number and angles of the radiation beams are chosen in advance and computers calculate how much dose may be delivered from each of the planned beams.
- the goal of IMRT is to increase the radiation dose to the areas that need it and reduce radiation exposure to specific sensitive areas of surrounding normal tissue.
- IGRT image-guided radiation therapy
- CT computed tomography
- MRI magnetic resonance imaging
- PET magnetic resonance imaging
- CT computed tomography
- MRI magnetic resonance imaging
- PET magnetic resonance imaging
- Imaging scans may be processed by computers to identify changes in a tumor's size and location due to treatment and to allow the position of the patient or the planned radiation dose to be adjusted during treatment as needed.
- Repeated imaging can increase the accuracy of radiation treatment and may allow reductions in the planned volume of tissue to be treated, thereby decreasing the total radiation dose to normal tissue.
- Tomotherapy is a type of image-guided IMRT.
- a tomotherapy machine is a hybrid between a CT imaging scanner and an external-beam radiation therapy machine.
- the part of the tomotherapy machine that delivers radiation for both imaging and treatment can rotate completely around the patient in the same manner as a normal CT scanner.
- Tomotherapy machines can capture CT images of the patient's tumor immediately before treatment sessions, to allow for very precise tumor targeting and sparing of normal tissue.
- Stereotactic radiosurgery can deliver one or more high doses of radiation to a small tumor.
- SRS uses extremely accurate image-guided tumor targeting and patient positioning.
- SRS can be used to treat small tumors with well-defined edges. It is most commonly used in the treatment of brain or spinal tumors and brain metastases from other cancer types. For the treatment of some brain metastases, patients may receive radiation therapy to the entire brain (called whole-brain radiation therapy) in addition to SRS. SRS requires the use of a head frame or other device to immobilize the patient during treatment to ensure that the high dose of radiation is delivered accurately.
- SBRT Stereotactic body radiation therapy
- SBRT delivers radiation therapy in fewer sessions, using smaller radiation fields and higher doses than 3D-CRT in most cases.
- SBRT may treat tumors that lie outside the brain and spinal cord. Because these tumors are more likely to move with the normal motion of the body, and therefore cannot be targeted as accurately as tumors within the brain or spine, SBRT is usually given in more than one dose.
- SBRT can be used to treat small, isolated tumors, including cancers in the lung and liver.
- SBRT systems may be known by their brand names, such as the CyberKnife®.
- Protons are a type of charged particle. Proton beams differ from photon beams mainly in the way they deposit energy in living tissue. Whereas photons deposit energy in small packets all along their path through tissue, protons deposit much of their energy at the end of their path (called the Bragg peak) and deposit less energy along the way. Use of protons may reduce the exposure of normal tissue to radiation, possibly allowing the delivery of higher doses of radiation to a tumor.
- Other charged particle beams such as electron beams may be used to irradiate superficial tumors, such as skin cancer or tumors near the surface of the body, but they cannot travel very far through tissue.
- brachytherapy Internal radiation therapy
- radiation sources radiation sources
- brachytherapy techniques are used in cancer treatment.
- Interstitial brachytherapy may use a radiation source placed within tumor tissue, such as within a prostate tumor.
- Intracavitary brachytherapy may use a source placed within a surgical cavity or a body cavity, such as the chest cavity, near a tumor.
- Episcleral brachytherapy which may be used to treat melanoma inside the eye, may use a source that is attached to the eye.
- radioactive isotopes can be sealed in tiny pellets or "seeds.” These seeds may be placed in patients using delivery devices, such as needles, catheters, or some other type of carrier. As the isotopes decay naturally, they give off radiation that may damage nearby cancer cells.
- Brachytherapy can be given as a low-dose-rate or a high-dose-rate treatment.
- low-dose- rate treatment cancer cells receive continuous low-dose radiation from the source over a period of several days.
- high-dose-rate treatment a robotic machine attached to delivery tubes placed inside the body may guide one or more radioactive sources into or near a tumor, and then removes the sources at the end of each treatment session.
- High-dose-rate treatment can be given in one or more treatment sessions.
- An example of a high-dose-rate treatment is the MammoSite® system.
- Bracytherapy may be used to treat patients with breast cancer who have undergone breast-conserving surgery.
- the drug ibritumomab tiuxetan may be used for the treatment of certain types of B-cell non-Hodgkin lymphoma (NHL).
- the antibody part of this drug recognizes and binds to a protein found on the surface of B lymphocytes.
- the combination drug regimen of tositumomab and iodine 1 T tositumomab (Bexxar®) may be used for the treatment of certain types of cancer, such as NHL.
- nonradioactive tositumomab antibodies may be given to patients first, followed by treatment with tositumomab antibodies that have 1 T attached.
- Tositumomab may recognize and bind to the same protein on B lymphocytes as ibritumomab.
- the nonradioactive form of the antibody may help protect normal B lymphocytes from being damaged by radiation from 1311.
- Some systemic radiation therapy drugs relieve pain from cancer that has spread to the bone (bone metastases). This is a type of palliative radiation therapy.
- the radioactive drugs samarium- 153-lexidronam (Quadramet®) and strontium-89 chloride (Metastron®) are examples of
- radiopharmaceuticals may be used to treat pain from bone metastases.
- Interferons are types of cytokines that occur naturally in the body. Interferon alpha, interferon beta, and interferon gamma are examples of interferons that may be used in cancer treatment.
- Colony-stimulating factors may also be used for the treatment of cancer.
- CSFs include, but are not limited to, G-CSF (filgrastim) and GM-CSF (sargramostim).
- CSFs may promote the division of bone marrow stem cells and their development into white blood cells, platelets, and red blood cells. Bone marrow is critical to the body's immune system because it is the source of all blood cells.
- CSFs may be combined with other anti-cancer therapies, such as chemotherapy.
- CSFs may be used to treat a large variety of cancers, including lymphoma, leukemia, multiple myeloma, melanoma, and cancers of the brain, lung, esophagus, breast, uterus, ovary, prostate, kidney, colon, and rectum.
- MOABs monoclonal antibodies
- MOABs monoclonal antibodies
- human cancer cells may be injected into mice.
- the mouse immune system can make antibodies against these cancer cells.
- the mouse plasma cells that produce antibodies may be isolated and fused with laboratory-grown cells to create "hybrid" cells called hybridomas.
- Hybridomas can indefinitely produce large quantities of these pure antibodies, or MOABs.
- MOABs may be used in cancer treatment in a number of ways. For instance, MOABs that react with specific types of cancer may enhance a patient's immune response to the cancer. MOABs can be programmed to act against cell growth factors, thus interfering with the growth of cancer cells.
- MOABs may be linked to other anti-cancer therapies such as chemotherapeutics,
- radioisotopes radioactive substances
- other biological therapies or other toxins.
- MOABs carrying radioisotopes may also prove useful in diagnosing certain cancers, such as colorectal, ovarian, and prostate.
- Rituxan® (rituximab) and Herceptin® (trastuzumab) are examples of MOABs that may be used as a biological therapy.
- Rituxan may be used for the treatment of non-Hodgkin lymphoma.
- Herceptin can be used to treat metastatic breast cancer in patients with tumors that produce excess amounts of a protein called HER2.
- MOABs may be used to treat lymphoma, leukemia, melanoma, and cancers of the brain, breast, lung, kidney, colon, rectum, ovary, prostate, and other areas.
- Cancer vaccines are another form of biological therapy. Cancer vaccines may be designed to encourage the patient's immune system to recognize cancer cells. Cancer vaccines may be designed to treat existing cancers (therapeutic vaccines) or to prevent the development of cancer (prophylactic vaccines). Therapeutic vaccines may be injected in a person after cancer is diagnosed. These vaccines may stop the growth of existing tumors, prevent cancer from recurring, or eliminate cancer cells not killed by prior treatments. Cancer vaccines given when the tumor is small may be able to eradicate the cancer. On the other hand, prophylactic vaccines are given to healthy individuals before cancer develops. These vaccines are designed to stimulate the immune system to attack viruses that can cause cancer. By targeting these cancer-causing viruses, development of certain cancers may be prevented.
- cervarix and gardasil are vaccines to treat human papilloma virus and may prevent cervical cancer.
- Therapeutic vaccines may be used to treat melanoma, lymphoma, leukemia, and cancers of the brain, breast, lung, kidney, ovary, prostate, pancreas, colon, and rectum. Cancer vaccines can be used in combination with other anti-cancer therapies.
- biological therapy includes nonspecific immunomodulating agents.
- Nonspecific immunomodulating agents are substances that stimulate or indirectly augment the immune system. Often, these agents target key immune system cells and may cause secondary responses such as increased production of cytokines and immunoglobulins.
- Two nonspecific immunomodulating agents used in cancer treatment are bacillus Calmette-Guerin (BCG) and levamisole.
- BCG may be used in the treatment of superficial bladder cancer following surgery. BCG may work by stimulating an inflammatory, and possibly an immune, response. A solution of BCG may be instilled in the bladder.
- Levamisole is sometimes used along with fluorouracil (5-FU) chemotherapy in the treatment of stage III (Dukes' C) colon cancer following surgery. Levamisole may act to restore depressed immune function.
- Photodynamic therapy is an anti-cancer treatment that may use a drug, called a photosensitizer or photosensitizing agent, and a particular type of light.
- a photosensitizer or photosensitizing agent When photos ensitizers are exposed to a specific wavelength of light, they may produce a form of oxygen that kills nearby cells.
- a photosensitizer may be activated by light of a specific wavelength. This wavelength determines how far the light can travel into the body. Thus, photos ensitizers and wavelengths of light may be used to treat different areas of the body with PDT.
- a photosensitizing agent may be injected into the bloodstream.
- the agent may be absorbed by cells all over the body but may stay in cancer cells longer than it does in normal cells. Approximately 24 to 72 hours after injection, when most of the agent has left normal cells but remains in cancer cells, the tumor can be exposed to light.
- the photosensitizer in the tumor can absorb the light and produces an active form of oxygen that destroys nearby cancer cells.
- PDT may shrink or destroy tumors in two other ways. The photosensitizer can damage blood vessels in the tumor, thereby preventing the cancer from receiving necessary nutrients. PDT may also activate the immune system to attack the tumor cells.
- the light used for PDT can come from a laser or other sources.
- Laser light can be directed through fiber optic cables (thin fibers that transmit light) to deliver light to areas inside the body.
- a fiber optic cable can be inserted through an endoscope (a thin, lighted tube used to look at tissues inside the body) into the lungs or esophagus to treat cancer in these organs.
- Other light sources include light-emitting diodes (LEDs), which may be used for surface tumors, such as skin cancer.
- PDT is usually performed as an outpatient procedure. PDT may also be repeated and may be used with other therapies, such as surgery, radiation, or chemotherapy.
- Extracorporeal photopheresis is a type of PDT in which a machine may be used to collect the patient's blood cells.
- the patient's blood cells may be treated outside the body with a photosensitizing agent, exposed to light, and then returned to the patient.
- ECP may be used to help lessen the severity of skin symptoms of cutaneous T-cell lymphoma that has not responded to other therapies.
- ECP may be used to treat other blood cancers, and may also help reduce rejection after transplants.
- photosensitizing agent such as porfimer sodium or Photofrin®
- Porfimer sodium may relieve symptoms of esophageal cancer when the cancer obstructs the esophagus or when the cancer cannot be satisfactorily treated with laser therapy alone.
- Porfimer sodium may be used to treat non-small cell lung cancer in patients for whom the usual treatments are not appropriate, and to relieve symptoms in patients with non-small cell lung cancer that obstructs the airways.
- Porfimer sodium may also be used for the treatment of precancerous lesions in patients with Barrett esophagus, a condition that can lead to esophageal cancer.
- Laser therapy may use high-intensity light to treat cancer and other illnesses.
- Lasers can be used to shrink or destroy tumors or precancerous growths.
- Lasers are most commonly used to treat superficial cancers (cancers on the surface of the body or the lining of internal organs) such as basal cell skin cancer and the very early stages of some cancers, such as cervical, penile, vaginal, vulvar, and non-small cell lung cancer.
- Laser therapy is often given through a flexible endoscope (a thin, lighted tube used to look at tissues inside the body).
- the endoscope is fitted with optical fibers (thin fibers that transmit light). It is inserted through an opening in the body, such as the mouth, nose, anus, or vagina. Laser light is then precisely aimed to cut or destroy a tumor.
- LITT Laser-induced interstitial thermotherapy
- interstitial laser photocoagulation also uses lasers to treat some cancers.
- LITT is similar to a cancer treatment called hyperthermia, which uses heat to shrink tumors by damaging or killing cancer cells.
- hyperthermia a cancer treatment
- LITT an optical fiber is inserted into a tumor. Laser light at the tip of the fiber raises the temperature of the tumor cells and damages or destroys them. LITT is sometimes used to shrink tumors in the liver.
- Laser therapy can be used alone, but most often it is combined with other treatments, such as surgery, chemotherapy, or radiation therapy.
- lasers can seal nerve endings to reduce pain after surgery and seal lymph vessels to reduce swelling and limit the spread of tumor cells.
- RNA-Seq analysis pipeline was employed on all samples (Fig. 5b).
- the analysis pipeline provided sequence quality metrics, filtering of contaminant reads, fragment size estimation, strand-specific library type estimation, spliced alignment of reads to the human reference genome (version hgl9/GrCh37), alignment performance metrics, generation of visualization tracks for genome browsers, and ab initio transcript assembly.
- the third-party tools used to process RNA- Seq data were selected based on computational performance, ease-of-use, user and community support, and experience.
- fragment size distribution for paired-end libraries
- fragment layout of each library was determined automatically by mapping a subset of the reads to a reference consisting of the 15,868 unique Ensembl v69 exons larger than 500bp that had no other overlapping features on either strand. These exons represent contiguous genomic regions where both paired-end reads from a single fragment could confidently be aligned. An alignment index was prepared from this reference using the bowtie-build utility.
- Alignment index files for Bowtie versions 0.12.8 and 2.0.2 were prepared from this reference using the -transcriptome index option in Tophat version 2.0.6.
- Sequence alignment metrics were computed using the Picard tools CollectMultipleMetrics and CollectRnaSeqMetrics.
- the Picard CollectRnaSeqMetrics diagnostic utility required gene annotation and ribosomal interval files as input.
- the "refFlat" table provided by the Illumina iGenomes download package (2012, March 9) was used as the gene annotation reference. Ribosomal DNA intervals were curated from the RepeatMasker table downloaded from the UCSC table browser (Karolchik, D. et al. Nucleic acids research 32, D493-496, (2004)). This table of repeat elements was originally provided for hgl9 by UCSC on 4/27/2009.
- transfrags ab initio assembled transcript fragments (transfrags) into a consensus transcriptome a bioinformatics method that (1) classifies and filters sources of background noise in individual libraries and (2) reassembles transfrags weighted by their expression levels from multiple libraries into a consensus transcriptome was utiized.
- RNA sequencing experiments that isolate poly-adenylated RNA from whole cells inadvertently capture variable amounts of incompletely processed RNA and genomic DNA4.
- These noise sequences manifest within ab initio transcript assemblies as intron retentions, mono-exonic intronic transfrags in the sense orientation, and relatively lowly expressed transfrags dispersed throughout intergenic regions (Cabili, M. N. et al. Genes & development 25, 1915-1927, (2011)).
- background noise complicates the correct assembly of mono-exonic transcripts, intronic transcripts, or both.
- the total unannotated sense-oriented intronic (intronic- like) transfrag population was used as a surrogate measure of both genomic and incompletely processed RNA levels, and the unannotated intergenic or antisense-oriented (intergenic-like) transfrag population as a surrogate measure of only genomic DNA levels. Comparing the transfrags in each category across all 6,503 libraries revealed significant variability in both the number and abundance of transfrags corresponding to noise (Fig. 6b). On average, intergenic-like transfrags constituted 8.6% of all transfrags (min: 0.65%, max: 43%), but only 0.88% of total FPKM per library (min: 0.16%, max: 16.8%).
- Intronic-like transfrags constituted 17% (min: 0.56%, max: 64%) of all transfrags and 2.0% (min: 0.18%, max: 54%) of total FPKM per library.
- bivariate kernel density estimates were converted using the abundance-recurrence axes separately for annotated and unannotated transfrags. These densities were mapped onto a square grid (50 x 50). The annotated density was then divided by the unannotated density at each grid point after adding a nominal value to avoid floating point overflow errors. This resulted in a new grid containing likelihood ratios for annotated versus unannotated transfrags along the abundance-recurrence axes. To account for the total noise present in the library, the likelihood estimates were weighted by the relative ratio of unannotated versus annotated transfrags in the library being classified.
- splicing graphs where nodes in the graph reflect contiguous exonic regions and edges correspond to splicing possibilities were generated (Fig. 7a).
- Nodes in the splicing graph are then pruned according to several criteria. First, low scoring ends in the graph that correspond to extraneously long exons or overhanging exons that extend into introns are removed. Second, nodes within introns are trimmed when their scores are less than a fraction of neighboring exons. Weakly connected components of the pruned splicing graphs are then extracted and processed independently.
- a splicing graph encompasses the milieu of possible isoforms that could be transcribed. Enumerating all possible paths through splicing graphs is impractical; many graphs have millions of paths of which only minute fractions are observed in vivo.
- the initial input transfrags provide partial paths through the splicing graph and also indicate which parts of the graph are more abundant.
- the approach described herein incorporates this partial path information by building a splicing pattern graph that subsumes the original splice graph (Fig. 7b).
- the splicing partem graph is a type of De Bruijn graph where each node represents a contiguous path of length k through the splice graph, and edges connect paths with k-1 nodes in common.
- each node in the graph carries a weight equal to the summed weights from all transcripts that share the node.
- the partial path length k is optimized to maximize the number of nodes in the path graph with the constraint that the summed node weights of transfrags with path length greater than or equal to k is above a userspecified fraction of the total score of all transfrags.
- This weight is allocated proportionally at nodes with multiple incoming or outgoing edges.
- This approach effectively extends all partial transcript fragments into full-length transcripts and assures that the sum of incoming and outgoing node weights at equivalent.
- a set of isoforms is predicted from the graph using a greedy algorithm. The algorithm finds and reports the highest abundance transcript by traversing the graph using dynamic programming. The weight of the transcript equals the minimum weight of all nodes in the path. The transcript weight is then subtracted from every node in the path and the dynamic programming procedure is repeated. Suboptimal transcripts are enumerated until a path weight falls below a fraction of the highest weighted transcript (e.g. the major isoform). The total number of isoforms produced from each gene can also be explicitly constrained. The meta- assembled isoforms are then reported in GTF and/or BED format. A genome track with summed node weights can optionally be reported in BedGraph format as well.
- AssemblyLine was developed as a software package written in Python and R to
- AssemblyLine accepts as input a set GTF files containing transfrags assembled from individual libraries. Transfrags of length less than 250bp were omitted from meta-assembly, and the remaining transfrags were labeled as 'annotated' or 'unannotated' relative to a reference GTF file (GENCODE version 16). An ab initio transfrag was considered 'annotated' if its exons overlapping any reference transcript exons on the identical strand. A recurrence score for each ab initio transfrag was computed as the average number of samples (replicate libraries from a single cell line or tissue were considered a single sample) per nucleotide with same-stranded transcription.
- Classification and filtering of 'background' and 'expressed' transfrags was performed by modeling the abundance (FPKM) and recurrence of 'annotated' and 'unannotated' transcripts using bivariate kernel density estimation on a square grid (grid size 50x50, bandwidth determined by Silverman's rule of thumb).
- a grid of likelihood ratios was derived from the 'annotated' and 'unannotated' grids by element-wise division at each grid point.
- the probability of each transfrag being 'annotated' was then determined by linearly interpolation onto this grid, and this probability was used as a surrogate measure for the probability that a transcript represented background noise.
- a likelihood ratio of less than or equal to one was used as a cutoff for filtering 'background' transcripts.
- Transcripts that overlapped a reference transcript on the same strand were designated annotated.
- a best match was chosen using the following criteria: (1) matching splicing pattern, (2) fraction of shared introns, and (3) fraction of shared transcribed bases.
- the biotype (protein, read-through, pseudogene, or lncRNA) for annotated transcripts was imputed from the best matching reference transcript.
- Annotated lncRNAs and unannotated transcripts were reclassified as either lncRNAs or TUCPs.
- Coding potential as predicted by integrating two sources of evidence: (1) predictions from the alignment-free Coding Potential Assessment Tool (CPAT) (Wang, L. et al. Nucleic acids research 41, e74, (2013)) and (2) searches for Pfam 27.0 matches (Finn, R. D. et al. Nucleic acids research 42, D222-230, (2014)).
- CPAT determines the coding probability of transcript sequences using a logistic regression model built from ORF size, Fickett TESTCODE statistic55, and hexamer usage bias.
- a CPAT probability cutoff was chosen by repeatedly randomly sampling 100,000 each of putative non- coding and protein-coding transcripts and optimizing on the balanced accuracy (average of sensitivity and specificity) metric (Fig. 9b,c).
- AUC average area-under-the-curve
- Thermo .raw files were obtained from the PRIDE database.
- Thermo .raw fiels were transformed into mzXML using
- Tandem output files were processed by PeptideProphet and ProteinProphet and for final output the data was filtered at peptide probability 0.5 and protein probability 0.9 to ensure protein FDR ⁇ 1%.
- transcripts were subjected to an additional confidence evaluation.
- IncRNAs in the MiTranscriptome were categorized into tiers based on their annotation status and the degree of matching of splice junctions to the reference annotation. Tier 1 transcripts are all annotated and tier 2 transcripts are unannotated.
- An empirical cumulative distribution function was developed by profiling the second highest expression value (across all 6,503 samples) for each tier 1 transcript. The second highest value was used to control for outlier expression. The second highest expression value for each tier 2 transcript was then fed into the distribution function to produce the confidence score.
- the housekeeping genes, CHMP2A, EMC 7, GPI, PSMB2, PSMB4, RAB7A, REEP5, SNRPD3 were used as loading controls56. Data was normalized first to housekeeping genes and then to the median value of all samples using the delta-delta Ct method and plotted as fold change over median. To ensure the specificity of the primers, 20 amplicons were further analyzed by Sanger sequencing.
- Cell lines and reagents All cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cell lines were maintained using standard conditions. Specifically, A549 were grown in F-12K plus 10% fetal bovine serum (FBS), LNCaP in RMPI1640 (Invitrogen) plus 10% FBS and 1% penicillin-streptomycin, and MCF7 in Eagle's Minimum Essential Media (EMEM) plus 10% FBS. All of the cell lines were grown at 37°C degrees in a 5% C02 cell culture incubator. To ensure identity, cell lines were genotyped at the University of Michigan Sequencing Core using
- STR Profiler Plus (Applied Biosystems) and compared with the short tandem repeat (STR) profiles of respective cell lines available in the STR Profile Database (ATCC). All of the cell lines were routinely tested and found to be free of Mycoplasma contamination.
- the Encode Project combined UW and Duke DNasel hypersensitivity regions were downloaded as a master file from EMBL-EBI, and filtered for any of the cell lines with H3k4me3 data. Peak enrichment files (BED format) were aggregated across all cell lines.
- TSSs Intervals of +/- 10 kilobases surrounding unique MiTranscriptome TSSs were generated using BEDTools 'slop' tool (Quinlan, A. R. & Hall, I. M. Bioinformatics 26, 841-842, (2010)). TSSs were filtered for expression in each cell line at RPKM>0.1.Basewise peak coverage was generated for each TSS interval using the BEDTools 'coverage' function and summarized across subsets of TSSs. Summed per-base coverage histograms were normalized by dividing by the number of expressed TSSs.
- transcripts in the assembly were studied using two metrics: (1) the fraction of significantly conserved bases (p ⁇ 0.01, phyloP algorithm), and (2) the maximally conserved 200nt sliding window (phastCons scores averaged within each window).
- the former captures independently conserved elements within a transcript regardless of position, and the latter captures contiguous regions of high conservation.
- the 200nt sliding window size was chosen to aid in discovery of putative ultraconserved elements (Bejerano, G. et al. Science 304, 1321-1325, (2004)).
- As a negative control the conservation of non-transcribed regions was measured using these metrics by randomly sampling contiguous length-matched intervals from intergenic and intronic space. Non-transcribed interval sampling was restricted to regions with valid 46-way conservation data.
- fractional basewise conservation and contiguous window conservation metrics were used to nominate highly conserved and ultraconserved transcripts, respectively. In both cases cutoffs for significant transcripts were determined by controlling the rate of observing elements with similar conservation levels within non-transcribed intergenic space at a level of 0.01. For fractional basewise conservation a score of 0.0947 (9.5% of transcript bases conserved at phyloP p-value ⁇ 0.01) corresponded to a false discovery rate ⁇ 0.01. At this cutoff the sensitivity for detecting protein- coding transcripts was 0.67. For contiguous sliding window conservation an average PhastCons probability of 0.9986 corresponded to a false discovery rate ⁇ 0.01.
- the sensitivity for detecting true positive ultraconserved non-coding elements downloaded from UCNEbase was 0.6926. Applying these criteria to the assembly yielded 6,034 IncRNAs (3.4%) and 541 TUCPs (4.7%) with significant basewise conservation levels. Additionally, 1 ,686 IncRNAs (0.96%) and 121 TUCPs (0.01%) harbored contiguous ultraconserved regions.
- GWAS SNPs were obtained from the National Human Genome Research Institute's GWAS catalog (Welter, D. et al. Nucleic acids research 42, D1001 -1006, (2014)). SNP haplotypes were excluded from the SNP overlap analysis, and a list of 1 1, 194 unique SNPs was obtained. The merged union of the RefSeq, UCSC, and GENCODE catalogs was used as a reference for comparison with MiTranscriptome.
- PhastCons algorithms for multiple alignments of 45 vertebrate genomes to the human genome were downloaded from the UCSC genome browser (Karolchik, D. et al. Nucleic acids research 42, D764- 770, (2014); Pollard, et al, Genome research 20, 1 10-121 , (2010); Siepel, A. et al. Genome research 15, 1034-1050, (2005)).
- the 'wigFix' formatted files were converting into 'bigWig' formatted files using the 'wigToBigWig' binary utility program provided by the UCSC genome browser (Karolchik et al., supra).
- For each transcript a vector of conservation scores for each exon was extracted using the 'bigWigToBedGraph' utility and concatenated into a single vector. Conservation metrics were then computed from these vectors.
- fracGW ASinstead of simply using nGW AS in this analysis is to control for the possibility that during the shuffle, transcripts could be shuffled into regions not represented on SNP arrays (e.g., regions unable to possess GWAS SNPs), falsely lowering the amount of GWAS SNP overlap by the shuffle. If transcripts are shuffled into regions that are not represented by the SNP background, both nGW AS and nbackground will decrease together, with fracGWAS relatively unchanged.
- Shuffling was performed using the BEDTools 'shuffle' tool.
- MiTranscriptome transcripts were grouped by transcription locus (e.g., regions of the genome that have contiguous transcription) prior to shuffling.
- Shuffling of transcript loci was performed to control for the fact that transcripts within a locus are spatially linked to one another. Shuffling without locus clustering would falsely elevate the amount of genome covered by transcripts, and subsequently elevate the number of SNPs overlapping the shuffled regions.
- MiTranscriptome proti en-coding transcripts was used as an exclusion file for these shuffles.
- FPKM Expression levels of the transcripts in the assembly were determined using Cufflinks (version 2.02 and 2.1.1)60. Normalized abundance estimates (FPKM) were computed for all MiTranscriptome transcripts, converted into approximate fragment count values, and aggregated into a matrix of expression data (Fig. 3a). Library size factors for expression normalization were computed by applying the geometric normalization method described by Anders and Huber (Genome biology 11, Rl 06 (2010)).
- SSEA Sample Set Enrichment Analysis
- the source code for this software is available online.
- the method adapts the weighted Kolmorgorov-Smirnoff (KS) tests proposed by Gene Set Enrichment Analysis (GSEA).
- GSEA Gene Set Enrichment Analysis
- SSEA tests for associations between individual gene expression observations (which could be transcript or gene expression) and sample sets.
- GSEA is analogous to performing GSEA on a 'transposed' input dataset.
- SSEA incorporates important features not provided by GSEA:
- KS-tests using normalized count data vectors as weights.
- the choice of power transformation influences the relative importance of precision versus recall during enrichment testing. For example, users aiming to discover genes new in molecular subtypes of a disease would prioritize precision over sensitivity, whereas a user aiming to discover ideal biomarkers may value sensitivity over precision.
- SSEA performs the weighted KS-test procedure described in GSEA28.
- the resulting enrichment score (ES) statistic describes the strength of association between the weights and the sample set.
- SSEA performs repeated enrichment tests using resampled count values to mimic observations from technical replicates and uses the median enrichment score (by default, 100 tests are performed).
- the basis for Poisson resampling as a legitimate model for technical replication was established by Marioni et al.62
- SSEA performs enrichment tests using randomly shuffled sample labels to derive a set of null enrichment scores with the same sign as the observed score (by default, 1000 null enrichment scores are computed).
- the nominal p value reported is the relative rank of the observed enrichment score within the null enrichment scores.
- SSEA maintains the null normalized enrichment score (NES) distributions for all transcripts in a sample set, and uses the null NES distribution to compute FDR q values in the same manner as proposed by Subramanian et al. (Proceedings of the National Academy of Sciences of the United States of America 102, 15545-15550, (2005)).
- NES null normalized enrichment score
- transcripts were ranked within each SSEA sample set by normalized enrichment score (NES) and assigned fractional ranks (e.g. a fractional rank of 0.95 implies the transcript ranked in the top 5th percentile of all transcripts in the sample set). Only significant results (FDR ⁇ le-7 for lineage analysis and FDR ⁇ le-3 for cancer versus normal analysis) were used. Unsupervised clustering was performed using Pearson correlation of log-transformed fractional ranks as a distance metric and Ward's method. Transcripts that were significantly associated with multiple sample sets were grouped with the most strongly associated sample set. Heatmaps were produced using the 'heatmap.2' function from the 'gplots' package in R. Guilt-by-association GSEA analysis
- RNA-Seq libraries were investigated by curating 7,256 poly-A+ RNA-Seq libraries from 25 independent studies, including 5,847 from TCGA, 928 from the Michigan Center for Translational Pathology (MCTP), 67 libraries from the Encyclopedia of DNA Elements (ENCODE), and 414 samples from other public datasets (Fig. 5a).
- An automated transcriptome assembly pipeline was developed and employed to process the raw sequencing datasets into ab initio transcriptome assemblies (Fig. 5b). This bioinformatics pipeline utilized approximately 1,870 core-months (average 0.26 core-months per library) on high-performance computing environments.
- RNA-Seq data constituted 493 billion fragments; individual libraries averaged 67.9M total fragments and 55.5M successful alignments to human chromosomes. On average 86% of aligned bases from individual libraries corresponded to annotated RefSeq exons, while the remaining 14% fell within introns or intergenic spacel5. Coarse quality control measures were used to account for variations in sequencing throughput, run quality, and RNA content by removing 753 libraries with (1) fewer than 20 million total fragments, (2) fewer than
- the set of 6,503 libraries passing quality control filters included 6,280 datasets from human tissues and 223 samples from cell lines. Of the tissue libraries, 5,298 originated from primary tumor specimens, 281 from metastases, and 701 from normal or benign adjacent tissues (Figs. 5e). This set of samples is referred to as the MiTranscriptome compendium.
- the libraries were partitioned into 18 cohorts by organ system (Fig. la), performed filtering and meta-assembly separately for each cohort, and re-merged the cohorts (Fig. lb).
- the individual ab initio assemblies collectively totaled -312M transcript predictions (transfrags) across all libraries.
- To perform filtering short transfrags ( ⁇ 250bp) and clipped short flanking exons ( ⁇ 15bp) were removed, leaving -304M transfrags (Fig. 6a). Whereas levels of annotated transfrags were relatively constant, fractions of unannotated intragenic and intergenic transcripts varied considerably across libraries (Fig. 6b).
- RNA-Seq experiments Two sources of background noise in RNA-Seq experiments that could give rise to unannotated mono-exonic transfrags are incompletely processed RNA and genomic DNA contamination (Fig. 6c). To minimize this noise, a conservative filtering scheme was used (Fig. 6d). 60M mono-exonic transfrags within introns that could have arisen from incompletely processed RNA were discarded. A machine learning method was developed to discriminate recurrent antisense and intergenic transcription from possible genomic DNA contamination.
- the approach models the empirical distributions of relative transcript abundance and recurrence (number of independent samples in which the transcript was observed) to determine optimal library-specific thresholds for distinguishing annotated from unannotated transcription.
- the classifier achieved remarkable performance (average AUC of 0.89, range 0.77-0.96) and displayed no bias for cancer versus normal samples (Fig. 6e).
- the classifier recovered test transcripts left out of the training process with 80% mean sensitivity (range 0.64-0.95, Fig. 6f).
- 3.2M of the 86.2M (3.7%) mono- exonic intergenic or antisense transfrags were retained for a total of 6.0M unannotated transfrags (6.75% of the original 89M).
- the filtered collection of 221M annotated and unannotated transfrags was subjected to meta-assembly.
- the meta-assembly algorithm first collapses transfrags into a splice graph and utilizes transcript abundance information to prune intron-retentions and trim long first or last exons (Fig. 7a).
- the algorithm integrates splicing pattern information by constructing a splicing pattern graph and traverses the graph using a greedy dynamic programming algorithm to generate full-length transcript predictions (Fig. 7b).
- meta-assembly of 7,471 transfrags in the chromosome 12 locus containing HOTAIR and HOXC11 produced just 17 transcripts, including transcripts that accurately matched annotated HOTAIR and HOXCl 1 isoforms (Fig. 7c).
- a consensus set of 384,066 predicted transcripts designated as the MiTranscriptome assembly was identified.
- Fig. lc were performed.
- increases in numbers of exons, splice sites, transcripts, and genes of 29%, 52%, 95%, and 57%, respectively, were observed relative to GENCODE, the most expansive of the three reference catalogs.
- the assembly was overlapped with a merged union of the three reference catalogs and the fraction of unannotated versus annotated transcripts were delineated for each cohort (Fig. 8a).
- the MiTranscriptome detected 94% and 93% of annotated RefSeq bases and splice sites, respectively. Detection of precise splicing patterns remains an ongoing challenge for in silico transcriptome reconstruction methods (Steijger, T. et al. Nature methods 10, 1177-1184, (2013)).
- transcripts were classified into one of five categories: (1) Protein-coding, (2) Read-through (implying a transcript overlapped multiple separate annotated genes), (3) Pseudogene, (4) IncRNA, and (5) Transcript of Unknown Coding Potential (TUCP) (Fig. 9a).
- the TUCP classification was originally described by Cabili et al. (supra) and pertains to long RNAs with features indicative of coding potential but not already annotated as protein coding. The ability to predict coding potential in silico using sequence features alone has important implications for ab initio transcript annotation studies.
- TUCPs were predicted by incorporating two methods: (1) predictions from the Coding Potential Assessment Tool (CPAT) (Wang, L. et al.
- transcripts overlapping annotated IncRNAs were flagged as TUCPs, indicating that previous annotation attempts may have identified incomplete ostensibly noncoding fragments that may actually comprise transcripts possessing robust ORFs.
- transcripts harboring a 418 amino acid ORF spanning 29 exons that overlapped three independent genes annotated by GENCODE as IncRNAs were identified, indicating that the annotated GENCODE IncRNAs may be incomplete partial annotations of a larger protein-coding gene (Fig. 2c).
- LncRNA and TUCP genes tended to have fewer exons than read-through or protein coding genes, but appreciable alternative splicing was observed for all classes of transcripts (Cabili et al, supra; Derrien, T. et al. Genome research 22, 1775-1789, (2012).) (Fig. 10a). Furthermore, it was observed that IncRNAs and TUCPs were expressed at lower levels than read-through or protein- coding transcripts, which is also consistent with previous studies (Prensner, J. R. et al. Nature biotechnology 29, 742-749, (2011)); Cabili et al, supra; Derrien et al, supra; Guttman, M. et al. Nature biotechnology 28, 503-510, (2010)) (Fig. 2d).
- TSS transcription start sites
- H3K4me3 histone 3 lysine 4 trimethylation
- ChlP-Seq ChlP-Seq
- RNA polymerase II (PolII) binding sites were compared.
- binding was only assessed for transcripts expressed in the cell lines being assayed, filtered TSSs for expression before intersection at a level of FPKM>0.1.
- LncRNA and TUCP promoters were enriched for these marks relative to randomly shuffled control regions, with maximal enrichment at the TSS (Fig. 2e-g).
- Enrichment was lower for IncRNA and TUCP promoters than for protein-coding genes, but much more enriched than pseudogenes, which may reflect their overall lower expression levels. These chromatin modification and polymerase binding data indicate that the assembled IncRNA and TUCP transcripts possess actively regulated promoters.
- MiTranscriptome contains only transcripts that have met this first-pass confidence evaluation.
- a confidence score (CS) system was developed. IncRNAs were classified into two tiers based on their annotation status and the matching of splice junctions, and a cumulative distribution function was built using the expression levels for the annotated IncRNAs (tier 1). The expression level of each unannotated lncRNA (tier 2) was then fed into the cumulative distribution function to calculate a CS for each lncRNA (Fig. 10b).
- the CS profile of the tier 1 and tier 2 transcripts was largely similar, with a slight enrichment in low confidence transcripts among the unannotated transcripts (e.g.
- IncRNAs The evolutionary conservation of IncRNAs has been a topic of ongoing conversation, with several reports indicating that IncRNAs are modestly conserved (Cabili et al, supra; Derrien et al. supra; Necsulea, A. et al. Nature 505, 635-640 (2014)).
- increases in both transcript and promoter conservation levels for IncRNAs and TUCPs relative to random control regions were observed (Fig. lOc-f). Shifts in the cumulative distributions of lncRNA and TUCP transcripts were greater for annotated transcripts relative to unannotated transcripts. This difference may reflect discovery bias favoring highly conserved genes detectable across multiple model systems.
- THCAT126 a previously unannotated intergenic IncRNA on chromosome 2q24, contains elements in its final exons that are conserved in nearly all vertebrates including zebrafish (Fig. 2i). Moreover, THCAT126 is expressed widely across many tissue types, and is expressed in multiple cancers, with a significant association in the thyroid cancer versus normal analysis (Fig. 2j). Highly conserved IncRNAs such as THCAT126 (and many other cancer-associated HICLINCs described below) provide an avenue for in vivo study of the role of IncRNAs in development and cancer.
- MiTranscriptome assembly was compared with 11,194 unique disease associated single nucleotide polymorphisms (SNPs) from a catalog of genome-wide association studies (GWAS) (Welter, D. et al. Nucleic acids research 42, D1001-1006, (2014)).
- SNPs single nucleotide polymorphisms
- MiTranscriptome transcripts overlapped 9,770 GWAS SNPs compared to just 7,050 SNPs overlapping GENCODE, UCSC, or RefSeq transcripts. Exonic overlap was 2,586 and 1,096 GWAS SNPs for the MiTranscriptome and aggregated reference catalogs, respectively (Fig. 13a,b).
- SSEA Sample Set Enrichment Analysis
- isoform- level expression data for the entire MiTranscriptome assembly was re-computed and samples from the compendia were grouped into fifty sample sets.
- a sample set represents a single condition for evaluating differential transcript expression.
- the sets in the present study included various cancer types (e.g., prostate cancers versus all other MiTranscriptome samples), normal tissues or cell types, and cancer versus normal comparisons within a single tissue type (e.g., prostate cancers versus benign prostate samples) (Fig. 3a).
- rsl3387042 downstream of a breast cancer SNP (rsl3387042) that has been implicated by six independent GWAS studies (Fig. 4f) (Li, J. et al. Breast cancer research and treatment 126, 717-727, (2011); Michailidou, K. et al. Nature genetics 45, 353-361, 361e351-352, (2013); Stacey, S. N. et al. Nature genetics 39, 865-869, (2007); Thomas, G. et al. Nature genetics 41, 579-584, doi: 10.1038/ng.353 (2009); Turnbull, C. et al. Nature genetics 42, 504-507, (2010)).
- the NHGRI GWAS catalog describes rsl3387042 as an intergenic SNP with no reported associated gene (Welter, D. et al.
- BRCAT49 provides a target for explaining the breast cancer association of this genomic region.
- cancer and lineage specificity support a role for BRCAT49 (and other similar cancer and lineage-specific IncRNAs) as a cancer specific
- MEAT6 Melanoma Associated Transcript-7
- MEAT6 Genomic investigation delineated MEAT6 as a partially annotated transcriptional variant of the UCSC IncRNA AK090788 IncRNA on chromosome 6q26 (Fig. 16a). However, MEAT6 utilizes an alternative start site and upstream exons absent from reference catalogs, highlighting the breadth and depth of transcriptome reconstruction effort. Expression of MEAT6 isoforms using the novel start site were highly specific to the melanoma samples in the MiTranscriptome cohort (Fig. 4e); however, isoforms lacking the MEAT6 start site had a dramatically different pan-cancer expression profile with almost no expression in melanoma (Fig. 16b).
- each transcript isoform was correlated to all annotated protein-coding genes for each relevant tissue cohort, and various cancer signatures were tested for enrichment with the most correlated or anti-correlated genes using the GSEA method.
- the gene sets were curated and categorized into cancer relevant categories: angiogenesis/hypoxia associated, metastasis associated, proliferation/cell-cycle associated, adhesion associated, DNA damage/repair associate, oncogenic association, and miscellaneous cancer association. In total over 14 thousand transcripts were analyzed with this method, and the significantly associated cancer gene sets are reported (Tables 2 and 3).
- VPS9D1-AS1.2 luad 3100 lncrna 400
Abstract
Provided herein are compositions and methods for cancer diagnosis, research and therapy, including but not limited to, cancer markers. In particular, provided herein are non-coding RNAs as diagnostic markers and clinical targets for cancer.
Description
NON-CODING RNAS AND USES THEREOF
The present Application claims priority to United States Provisional Patent Application Serial
Number 62/088,772 filed December 8, 2014, the disclosure of which is herein incorporated by reference in its entirety.
STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH OR DEVELOPMENT
This invention was made with government support under CA111275, CA154365 and CA069568 awarded by the National Institutes of Health and support under W81XWH-11-1-0337 awarded by the Army / MRMC. The Government has certain rights in the invention.
FIELD OF THE DISCLOSURE
Provided herein are compositions and methods for cancer diagnosis, research and therapy, including but not limited to, cancer markers. In particular, provided herein are non-coding RNAs as diagnostic markers and clinical targets for cancer.
BACKGROUND OF THE DISCLOSURE
Afflicting one out of nine men over age 65, prostate cancer (PCA) is a leading cause of male cancer-related death, second only to lung cancer (Abate-Shen and Shen, Genes Dev 14:2410 [2000]; Ruijter et al , Endocr Rev, 20:22 [1999]). The American Cancer Society estimates that about 184,500 American men will be diagnosed with prostate cancer and 39,200 will die in 2001.
Prostate cancer is typically diagnosed with a digital rectal exam and/or prostate specific antigen (PSA) screening. An elevated serum PSA level can indicate the presence of PCA. PSA is used as a marker for prostate cancer because it is secreted only by prostate cells. A healthy prostate will produce a stable amount ~ typically below 4 nanograms per milliliter, or a PSA reading of "4" or less ~ whereas cancer cells produce escalating amounts that correspond with the severity of the cancer. A level between 4 and 10 may raise a doctor's suspicion that a patient has prostate cancer, while amounts above 50 may show that the tumor has spread elsewhere in the body.
When PSA or digital tests indicate a strong likelihood that cancer is present, a transrectal ultrasound (TRUS) is used to map the prostate and show any suspicious areas. Biopsies of various sectors of the prostate are used to determine if prostate cancer is present. Treatment options depend on the stage of the cancer. Men with a 10-year life expectancy or less who have a low Gleason number and whose tumor has not spread beyond the prostate are often treated with watchful waiting (no treatment). Treatment options for more aggressive cancers include surgical treatments such as radical prostatectomy (RP), in which the prostate is completely removed (with or without nerve sparing techniques) and radiation, applied through an external beam that directs the dose to the prostate from outside the body or via low-dose radioactive seeds that are implanted within the prostate to kill cancer cells locally. Anti-androgen hormone therapy is also used, alone or in
conjunction with surgery or radiation. Hormone therapy uses luteinizing hormone-releasing hormones (LH-RH) analogs, which block the pituitary from producing hormones that stimulate testosterone production. Patients must have injections of LH-RH analogs for the rest of their lives.
While surgical and hormonal treatments are often effective for localized PCA, advanced disease remains essentially incurable. Androgen ablation is the most common therapy for advanced PCA, leading to massive apoptosis of androgen-dependent malignant cells and temporary tumor regression. In most cases, however, the tumor reemerges with a vengeance and can proliferate independent of androgen signals.
The advent of prostate specific antigen (PSA) screening has led to earlier detection of PCA and significantly reduced PCA-associated fatalities. However, the impact of PSA screening on cancer-specific mortality is still unknown pending the results of prospective randomized screening studies (Etzioni et al , J. Natl. Cancer Inst, 91 : 1033 [1999]; Maattanen et al , Br. J. Cancer 79: 1210 [1999]; Schroder et al, J. Natl. Cancer Inst, 90: 1817 [1998]). A major limitation of the serum PSA test is a lack of prostate cancer sensitivity and specificity especially in the intermediate range of PSA detection (4-10 ng/ml). Elevated serum PSA levels are often detected in patients with non-malignant conditions such as benign prostatic hyperplasia (BPH) and prostatitis, and provide little information about the aggressiveness of the cancer detected. Coincident with increased serum PSA testing, there has been a dramatic increase in the number of prostate needle biopsies performed (Jacobsen et al , JAMA 274: 1445 [1995]). This has resulted in a surge of equivocal prostate needle biopsies (Epstein and Potter J. Urol, 166:402 [2001]). Thus, development of additional serum and tissue biomarkers to supplement PSA screening is needed.
SUMMARY OF THE DISCLOSURE
Provided herein are compositions and methods for cancer diagnosis, research and therapy, including but not limited to, cancer markers. In particular, provided herein are non-coding RNAs as diagnostic markers and clinical targets for cancer.
In some embodiments, the present disclosure provides a method of screening for the presence of cancer in a subject, comprising (a) contacting a biological sample from a subject with a gene expression detection assay, wherein said gene expression detection assay comprises a gene expression informative reagent for identification of the level of expression of one or more non- coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309; (b) detecting the level of expression of said non-coding in said sample using an in vitro assay; and (c) diagnosing cancer in said subject when an increased level of expression of said non-coding RNAs in said sample relative to the level in normal cells is detected. In some embodiments, the RNAs are converted to cDNA prior to or during detection. In some embodiments, the sample is selected from,
for example, tissue, blood, plasma, serum, urine, urine supernatant, urine cell pellet, semen, prostatic secretions or prostate cells. In some embodiments, the detection is carried out utilizing a method selected from, for example, a sequencing technique, a nucleic acid hybridization technique, or a nucleic acid amplification technique. In some embodiments, the nucleic acid amplification technique is selected from, for example, polymerase chain reaction, reverse transcription polymerase chain reaction, transcription-mediated amplification, ligase chain reaction, strand displacement amplification, or nucleic acid sequence based amplification. The present disclosure is not limited to a particular cancer. Examples include, but are not limited to, prostate cancer, breast cancer, acute myeloid leukemia (AML), chronic myeloid leukemia (CML), myeloproliferative neoplasia (MPN)), lower grade glioma (LGG), glioblastome multiforme (GBM)), cervical cancer, head and neck cancer, lung squamous cell cancer, lung adenocarcinoma, kidney cancer, papillary cell carcimona, or bladder cancer. In some embodiments, the reagent is a pair of amplification oligonucleotides, a sequencing primer, or an oligonucleotide probe. In some embodiments, the reagent comprises one or more labels. In some embodiments, the one or more non-coding RNAs is two or more (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 50, 75, 100, or more).
Further embodiments provide a method of identifying gene expression associated with cancer, comprising (a) contacting a biological sample from a subject with a gene expression detection assay, wherein said gene expression detection assay comprises a gene expression informative reagent for identification of the level of expression of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1 -2309; (b) detecting the level of expression of said non-coding RNA in said sample using an in vitro assay; and (c) identifying gene expression subjects at risk of prostate cancer metastasis when an increased level of expression of said non-coding RNA said sample relative to the level in normal prostate cells is detected.
Additonal embodiments provide a system for analyzing a cancer, comprising: a probe set comprising a plurality of probes, wherein the plurality of probes comprises a sequence that hybridizes to at least a portion of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309 or the corresponding cDNA; and a computer model or algorithm for analyzing an expression level and/or expression profile of said non-coding RNA hybridized to the probe in a sample from a subject. In some embodiments, the system further comprises one or more of computer memory for capturing and storing an expression profile, a computer-processing device, optionally connected to a computer network, a software module executed by the computer-processing device to analyze an expression profile, a software module executed by the computer-processing device to compare the expression profile to a standard or
control, a software module executed by the computer-processing device to determine the expression level of the non-coding RNA, a software module executed by the computer-processing device to transmit an analysis of the expression profile to the subject or a medical professional treating the subject or a software module executed by the computer-processing device to transmit a diagnosis or prognosis to the subject or a medical professional treating the subject.
Also provided is a probe set for assessing a cancer status of a subject comprising a plurality of probes, wherein the probes in the probe set are capable of detecting an expression level of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1- 2309 or the corresponding cDNA.
Yet other embodiments provide a composition comprising one or more reaction mixtures, wherein each reaction mixture comprises a complex of a non-coding RNAs selected from those described by SEQ ID NOs: 1-2309 or the corresponding cDNA and a probe that binds to said non- coding RNA.
Additionaly provided herein are methods of treating cancer, comprising contacting a cancer cell with a compound (e.g., siRNA or antisense oligonucleotide) that specifically targets one or more non-coding RNAs selected from those described by SEQ ID NOs: 1-2309. In some embodiments, the cell is in a subject.
Additional embodiments are described herein.
DESCRIPTION OF THE FIGURES
Figure 1 shows that^4Z> initio transcriptome assembly reveals an expansive landscape of human transcription, (a) Pie chart showing composition and cohort sizes for RNA-Seq transcriptome reconstruction, (b) Workflow diagram for transcriptome reconstruction, (c) Bar chart comparing exons, splice sites, transcripts, and genes in the MiTranscriptome assembly with the RefSeq (Dec, 2013), UCSC (Dec, 2013) and GENCODE (release 19) catalogs.
Figure 2 shows characterization of the MiTranscriptome assembly, (a) Pie chart of composition and quantities of IncRNA, transcripts of unknown coding potential (TUCP), expressed pseudogene, read-through, and protein-coding genes in the MiTranscriptome assembly, (b) Pie charts of number of IncRNAs and TUCP genes (top) unannotated versus annotated relative to reference catalogs and (bottom) intragenic versus intergenic. (c) Genomic view of the chromosome 16pl3.3 locus, (d) Empirical cumulative distribution plot comparing the maximum expression (FPKM) of the major isoform of each gene across gene categories, (e, f, and g) Plots of enrichment of lOkb intervals surrounding expressed transcription start sites (TSSs
with RPM>0.1) with aggregated ENCODE data from 13 cell lines for (e) H3K4me3 ChlP-Seq, (f) PolII transcription factor binding sites, and (g) DNase hypersensitivity, (h) Scatter plot with marginal
histograms depicting the distribution of full transcript conservation levels (x axis) and maximal 200bp window conservation levels (y axis) for IncRNA and TUCP transcripts.
Figure 3 shows exemplary methodology for discovering cancer-associated IncRNAs. (a) Transcript expression analysis workflow, (b) Heatmap showing concordance of SSEA algorithm with cancer gene signatures obtained from the Oncomine database, (c) Sample set enrichment density plots showing spectrum of transcript enrichment scores (ES) obtained from SSEA analysis of breast carcinomas versus corresponding normal samples, (d and e) SSEA enrichment plots and expression boxplots for the IncRNAs (d) HOTAIR and (e) MEG3. (f) Sample set enrichment density plots showing spectrum of transcript enrichment scores (ES) obtained from SSEA analysis of prostate carcinomas versus corresponding normal samples, (g and h) Transcript enrichment bar plots for prostate cancer-specific IncRNAs (g) PCA3 and (h) SChLAPl across Cancer vs. Normal, Cancer Type and Normal Type sample sets.
Figure 4 shows discovery of lineage-associated and cancer-associated IncRNAs in the MiTranscriptome compendia, (a) Heatmap of lineage-specific IncRNAs. (b) Heatmap of cancer- specific IncRNAs nominated by SSEA Cancer vs. Normal analysis of 12 cancer types (columns), (c) Scatter plots showing enrichment score for Cancer vs. Normal (x axis) and Cancer Lineage (y axis) for all lineage-specific and cancer-associated IncRNA transcripts across 12 cancer types, (d) Boxplot comparing the performance of cancer- and lineage-associated IncRNAs corresponding to unannotated or annotated IncRNAs or protein-coding transcripts (including readthroughs) across 12 cancer types, (e) Expression data for MEAT6 across all MiTranscriptome cancer and normal tissue type cohorts, (f) Genomic view of chromosome 2q35 locus, (g) Expression data for BRCAT49 across all MiTranscriptome cancer and normal tissue type cohorts.
Figure 5 shows curation and processing of samples in the MiTrascriptome compendia. A. Pie chart showing thenumber of studies curated from TCGA, ENCODE, MCTP, and other datasets.
Figure 6 shows transfrag filtering, a) Pie chart showing the number of studies curated from datasets. b) workflow for bioinformatics processing of individual RNA-SEQ libraries. C) scatter plot showing total fragments (x-axis) and the fraction of aligned fragments (y-axis for each RNA-SEQ library. D) dot plot showing the fraction of aligned bases corresponding to mRNA, intronic regions, or intergenic regions. EOpie chart showing numbers of primare tumors, metastatic tumors, benign adjacent tissues or healthy tissues, or cell lines for RNA-SEQ libraries.
Figure 7 shows meta assembly, a, schematic of transcriptome meta-assembly algorithm using a simplified example with three transtrags transcribed from left to right, b, The pruned splice graph from panel a is subjected to meta-assembly. c, Genome view showing an example of the meta- assembly procedure for breast cohort transfrags in a chromosome 12ql3.3 locus containing the
LncRNA HOTAIR and the protein-coding gene HOXC11 on opposite strands (chrl2:54,349,995- 54,377,376, hgl9).
Figure 8 shows characterization of unannotated transcripts, a, Bar plots comparing numbers of unannolaled versus different dasses of annotated transcripts for each of the 18 cohorts, b, 001 plots depicting comparison of MiTransriptome with reference transcripts from RefSeq, UCSC, or GENCODE. c, Dot plots comparing the basewise, splice site, and splicing partem precision and sensitivity of MiTranscriptome and GENCODE using RefSeq (left) or Cabili et al. LncRNAs (right)
Figure 9 shows classification of transcripts of unknown coding potential, a, Decision tree showing categorization of ab initio transcripts, b, ROC curve comparing false positive rate (x axis) with true positive rate (yaxis) for CPAT coding potential predictions of ncRNAs versus protein- coding genes, c, Curve comparing probability cutoff (x axis) with balanced accuracy (yaxis). d, Scatter plot comparing frequencies of Pfam domain occurrences in non-transcribed intergenic space versus transcribed regions, e, Three-dimensional scatter plot comparing Fickett score (x axis), ORF size (yaxis), and Hexamer score (z axis) for all transcripts. f,g,h Boxplots comparing ORF size (f), Hexamer score (g), and Fickett score (h).
Figure 10 shows Mitranscriptome characterization, a, Comparison of the relationship of maximum number of exons per gene to the number of isoforms per gene, b, Density histogram depicting the confidence scores for annotated and unannotated IncRNAs. c, Cumulative distribulion plot for basewise conservation fraction of proteins, read-throughs, pseudogenes, TUCPs, IncRNAs. d, Bar plot showing KS test statistics for classes of transcripts versus random intergenic controls, e, Cumulative dislribution plot for promoter conservation (legend shared with a), f, Bar plot showing KS tests for promoter conservation versus random intergenic regions, g, ROC curve for predicting conservation of protein coding genes versus random intergenic controls.
Figure 11 shows validation of lncRNA transcripts.
Figure 12 shows validation of lncRNA transcripts, a, b, Representative example of two of twenty previously unannotated lncRNA transcripts that were analyzed by Sanger sequencing to ensure primer specificity with their associated chromatograms. c, Heatmap representation of the correlation between qPCR (fold change over median) with RNA-seq (FPKM) of 100 selected transcripts in cell lines A549, LNCaP, and MCF7.
Figure 13 shows enrichment of MiTranscriptome assembly for disease-associated regions, a,
Venn diagram comparing coverage of disease or trait associated genomic regions for the
MiTranscriptome assembly in comparison to reference catalog, b, Pie charts comparing distributions of intronic and exonic GWAS SNP coverage of the MiTranscriptome assembly (left) and reference
catalogs (right), c, Dot plot showing enrichment of GWAS SNPs (cirde) versus random SNPs (diamond) for novel intergenic IncRNAs and TUCPs.
Figure 14 shows discovery of lineage associated and cancer associated transcripts, a, Heatmap of lineage-specific transcripts (LATs) nominated by SSEA. b, Heatmap of cancer-specific transcripts (CATS) nominated by SSEA.
Figure 15 shows lineage-specific and cancer-specific transcripts, a, Scatter plot grid showing lineage-specific and cancer-specific transcripts (CLATs) nominated by SSEA. b and c, Boxplots comparing the perfonnance of (b) positively enriched CLATs and c) negatively enriched CLATs for each transcript category across 12 cancer types.
Figure 16 shows examples of cancer and/or lineage associated transcripts), a, Genomic view of chromosome 6q26-q271ocus. b, Expression data for MEAT6 (demarcated by asterisk in a).
Expression profile for cancer and lineage assodated transcripts across all MiTraoscriptome tissue cohorts are shown for c, lung adenocarcinoma, and d, thyroid cancer
DEFINITIONS
To facilitate an understanding of the present disclosure, a number of terms and phrases are defined below:
As used herein, the terms "detect", "detecting" or "detection" may describe either the general act of discovering or discerning or the specific observation of a composition. Detecting a
composition may comprise determining the presence or absence of a composition. Detecting may comprise quantifying a composition. For example, detecting comprises determining the expression level of a composition. The composition may comprise a nucleic acid molecule. For example, the composition may comprise at least a portion of the ncRNAs disclosed herein. Alternatively, or additionally, the composition may be a detectably labeled composition.
As used herein, the term "subject" refers to any organisms that are screened using the diagnostic methods described herein. Such organisms preferably include, but are not limited to, mammals (e.g., murines, simians, equines, bovines, porcines, canines, felines, and the like), and most preferably includes humans. Alternatively, the organism is an avian, amphibian, reptile or fish.
The term "diagnosed," as used herein, refers to the recognition of a disease by its signs and symptoms, or genetic analysis, pathological analysis, histological analysis, and the like.
As used herein, the term "characterizing cancer in a subject" refers to the identification of one or more properties of a cancer sample in a subject, including but not limited to, the presence of benign, pre-cancerous or cancerous tissue, the stage of the cancer, and the subject's prognosis.
Cancers may be characterized by the identification of the expression of one or more cancer marker genes, including but not limited to, the ncRNAs disclosed herein.
As used herein, the term "stage of cancer" refers to a qualitative or quantitative assessment of the level of advancement of a cancer. Criteria used to determine the stage of a cancer include, but are not limited to, the size of the tumor and the extent of metastases (e.g., localized or distant).
As used herein, the term "nucleic acid molecule" refers to any nucleic acid containing molecule, including but not limited to, DNA or RNA. The nucleic acid molecule may comprise one or more nucleotides. The term encompasses sequences that include any of the known base analogs of DNA and RNA including, but not limited to, 4-acetylcytosine, 8-hydroxy-N6-methyladenosine, aziridinylcytosine, pseudoisocytosine, 5-(carboxyhydroxylmethyl) uracil, 5-fluorouracil,
5-bromouracil, 5-carboxymethylaminomethyl-2-thiouracil, 5-carboxymethylaminomethyluracil, dihydrouracil, inosine, N6-isopentenyladenine, 1 -methyladenine, 1-methylpseudouracil,
1- methylguanine, 1 -methylinosine, 2,2-dimethylguanine, 2-methyladenine, 2-methylguanine, 3- methylcytosine, 5-methylcytosine, N6-methyladenine, 7-methylguanine,
5-methylaminomethyluracil, 5-methoxyaminomethyl-2-thiouracil, beta-D-mannosylqueosine, 5'-methoxycarbonylmethyluracil, 5 -methoxy uracil, 2-methylthio-N6-isopentenyladenine, uracil-5-oxyacetic acid methylester, uracil-5-oxyacetic acid, oxybutoxosine, pseudouracil, queosine,
2- thiocytosine, 5-methyl-2-thiouracil, 2-thiouracil, 4-thiouracil, 5-methyluracil, N-uracil-5-oxyacetic acid methylester, uracil-5-oxyacetic acid, pseudouracil, queosine, 2-thiocytosine, and
2,6-diaminopurine.
The term "gene" refers to a nucleic acid (e.g. , DNA) sequence that comprises coding sequences necessary for the production of a polypeptide, precursor, or RNA (e.g., rRNA, tRNA). The polypeptide can be encoded by a full length coding sequence or by any portion of the coding sequence so long as the desired activity or functional properties (e.g., enzymatic activity, ligand binding, signal transduction, immunogenicity, etc.) of the full-length or fragments are retained. The term also encompasses the coding region of a structural gene and the sequences located adjacent to the coding region on both the 5' and 3' ends for a distance of about 1 kb or more on either end such that the gene corresponds to the length of the full-length mRNA. Sequences located 5' of the coding region and present on the mRNA are referred to as 5' non-translated sequences. Sequences located 3' or downstream of the coding region and present on the mRNA are referred to as 3' non-translated sequences. The term "gene" encompasses both cDNA and genomic forms of a gene. A genomic form or clone of a gene contains the coding region interrupted with non-coding sequences termed "introns" or "intervening regions" or "intervening sequences." Introns are segments of a gene that are transcribed into nuclear RNA (hnRNA); introns may contain regulatory elements such as enhancers. Introns are removed or "spliced out" from the nuclear or primary transcript; introns
therefore are absent in the messenger RNA (mRNA) transcript. The mRNA functions during translation to specify the sequence or order of amino acids in a nascent polypeptide.
As used herein, the term "oligonucleotide," refers to a short length of single-stranded polynucleotide chain. Oligonucleotides are typically less than 200 residues long (e.g. , between 15 and 100), however, as used herein, the term is also intended to encompass longer polynucleotide chains. Oligonucleotides are often referred to by their length. For example a 24 residue
oligonucleotide is referred to as a "24-mer". Oligonucleotides can form secondary and tertiary structures by self-hybridizing or by hybridizing to other polynucleotides. Such structures can include, but are not limited to, duplexes, hairpins, cruciforms, bends, and triplexes.
As used herein, the terms "complementary" or "complementarity" are used in reference to polynucleotides (i.e. , a sequence of nucleotides) related by the base-pairing rules. For example, the sequence "5'-A-G-T-3'," is complementary to the sequence "3'-T-C-A-5\" Complementarity may be "partial," in which only some of the nucleic acids' bases are matched according to the base pairing rules. Or, there may be "complete" or "total" complementarity between the nucleic acids. The degree of complementarity between nucleic acid strands has significant effects on the efficiency and strength of hybridization between nucleic acid strands. This is of particular importance in amplification reactions, as well as detection methods that depend upon binding between nucleic acids.
The term "homology" refers to a degree of complementarity. There may be partial homology or complete homology (i.e., identity). A partially complementary sequence is a nucleic acid molecule that at least partially inhibits a completely complementary nucleic acid molecule from hybridizing to a target nucleic acid is "substantially homologous." The inhibition of hybridization of the completely complementary sequence to the target sequence may be examined using a hybridization assay (Southern or Northern blot, solution hybridization and the like) under conditions of low stringency. A substantially homologous sequence or probe will compete for and inhibit the binding (i.e. , the hybridization) of a completely homologous nucleic acid molecule to a target under conditions of low stringency. This is not to say that conditions of low stringency are such that nonspecific binding is permitted; low stringency conditions require that the binding of two sequences to one another be a specific (i.e. , selective) interaction. The absence of non-specific binding may be tested by the use of a second target that is substantially non-complementary (e.g. , less than about
30% identity); in the absence of non-specific binding the probe will not hybridize to the second non- complementary target.
As used herein, the term "hybridization" is used in reference to the pairing of complementary nucleic acids. Hybridization and the strength of hybridization (i.e. , the strength of the association
between the nucleic acids) is impacted by such factors as the degree of complementary between the nucleic acids, stringency of the conditions involved, the Tm of the formed hybrid, and the G:C ratio within the nucleic acids. A single molecule that contains pairing of complementary nucleic acids within its structure is said to be "self-hybridized."
As used herein the term "stringency" is used in reference to the conditions of temperature, ionic strength, and the presence of other compounds such as organic solvents, under which nucleic acid hybridizations are conducted. Under "low stringency conditions" a nucleic acid sequence of interest will hybridize to its exact complement, sequences with single base mismatches, closely related sequences (e.g. , sequences with 90% or greater homology), and sequences having only partial homology (e.g. , sequences with 50-90% homology). Under 'medium stringency conditions," a nucleic acid sequence of interest will hybridize only to its exact complement, sequences with single base mismatches, and closely relation sequences (e.g. , 90% or greater homology). Under "high stringency conditions," a nucleic acid sequence of interest will hybridize only to its exact complement, and (depending on conditions such a temperature) sequences with single base mismatches. In other words, under conditions of high stringency the temperature can be raised so as to exclude hybridization to sequences with single base mismatches.
The term "isolated" when used in relation to a nucleic acid, as in "an isolated
oligonucleotide" or "isolated polynucleotide" refers to a nucleic acid sequence that is identified and separated from at least one component or contaminant with which it is ordinarily associated in its natural source. Isolated nucleic acid is such present in a form or setting that is different from that in which it is found in nature. In contrast, non-isolated nucleic acids as nucleic acids such as DNA and RNA found in the state they exist in nature. For example, a given DNA sequence (e.g. , a gene) is found on the host cell chromosome in proximity to neighboring genes; RNA sequences, such as a specific mRNA sequence encoding a specific protein, are found in the cell as a mixture with numerous other mRNAs that encode a multitude of proteins. However, isolated nucleic acid encoding a given protein includes, by way of example, such nucleic acid in cells ordinarily expressing the given protein where the nucleic acid is in a chromosomal location different from that of natural cells, or is otherwise flanked by a different nucleic acid sequence than that found in nature. The isolated nucleic acid, oligonucleotide, or polynucleotide may be present in single-stranded or double-stranded form. When an isolated nucleic acid, oligonucleotide or polynucleotide is to be utilized to express a protein, the oligonucleotide or polynucleotide will contain at a minimum the sense or coding strand (i.e. , the oligonucleotide or polynucleotide may be single-stranded), but may contain both the sense and anti-sense strands (i.e. , the oligonucleotide or polynucleotide may be double-stranded).
As used herein, the term "purified" or "to purify" refers to the removal of components (e.g., contaminants) from a sample. For example, antibodies are purified by removal of contaminating non-immunoglobulin proteins; they are also purified by the removal of immunoglobulin that does not bind to the target molecule. The removal of non-immunoglobulin proteins and/or the removal of immunoglobulins that do not bind to the target molecule results in an increase in the percent of target-reactive immunoglobulins in the sample. In another example, recombinant polypeptides are expressed in bacterial host cells and the polypeptides are purified by the removal of host cell proteins; the percent of recombinant polypeptides is thereby increased in the sample.
The term "label" as used herein refers to any atom or molecule that can be used to provide a detectable (preferably quantifiable) effect, and that can be attached to a nucleic acid or protein. Labels include but are not limited to dyes; radiolabels such as 2P; binding moieties such as biotin; haptens such as digoxgenin; luminogenic, phosphorescent or fluorogenic moieties; and fluorescent dyes alone or in combination with moieties that can suppress or shift emission spectra by
fluorescence resonance energy transfer (FRET). Labels may provide signals detectable by fluorescence, radioactivity, colorimetry, gravimetry, X-ray diffraction or absorption, magnetism, enzymatic activity, and the like. A label may be a charged moiety (positive or negative charge) or alternatively, may be charge neutral. Labels can include or consist of nucleic acid or protein sequence, so long as the sequence comprising the label is detectable. In some embodiments, nucleic acids are detected directly without a label (e.g., directly reading a sequence).
As used herein, the term "sample" is used in its broadest sense. In one sense, it is meant to include a specimen or culture obtained from any source, as well as biological and environmental samples. Biological samples may be obtained from animals (including humans) and encompass fluids, solids, tissues, and gases. Biological samples include blood products, such as plasma, serum and the like. Such examples are not however to be construed as limiting the sample types applicable to the present disclosure.
DETAILED DESCRIPTION OF THE DISCLOSURE
Provided herein are compositions and methods for cancer diagnosis, research and therapy, including but not limited to, cancer markers. In particular, provided herein are non-coding RNAs as diagnostic markers and clinical targets for cancer.
Many RNA transcripts are not classical protein-coding genes. There is an abundance of unknown, uncharacterized RNA species in the human transcriptome (e.g., lncRNA or long non- coding RNAs). Provided herein are compositions and methods for utilizing such non-coding RNAs in diagnostic, research, and screening methods.
I. Diagnostic and Screening Methods
As described herein, embodiments of the present disclosure provide diagnostic and screening methods that utilize the detection of one or more non-coding RNAs. Exemplary non-coding RNAs include, but are not limited to, those described in SEQ ID NOs: 1-2309. Exemplary, non-limiting methods are described herein.
Any patient sample suspected of containing the non-coding RNAs may be tested according to methods of embodiments of the present disclosure. By way of non-limiting examples, the sample may be tissue (e.g. , a biopsy sample, a prostate biopsy sample or a tissue sample obtained by prostatectomy), blood, urine, semen, prostatic secretions or a fraction thereof (e.g., plasma, serum, urine supernatant, urine cell pellet, cells or prostate cells). A urine sample may be collected immediately following an attentive digital rectal examination (DRE), which causes prostate cells from the prostate gland to shed into the urinary tract.
In some embodiments, the patient sample is subjected to preliminary processing designed to isolate or enrich the sample for the non-coding RNAs or cells that contain the non-coding RNAs. A variety of techniques known to those of ordinary skill in the art may be used for this purpose, including but not limited to: centrifugation; immunocapture; cell lysis; nucleic acid amplification; and, nucleic acid target capture (See, e.g., EP Pat. No. 1 409 727, herein incorporated by reference in its entirety).
The non-coding RNAs may be detected along with other markers in a multiplex or panel format. Markers may be selected for their predictive value alone or in combination with non-coding RNAs described herein (e.g., one or more of SEQ ID NOs: 1-2309). Exemplary prostate cancer markers include, but are not limited to: AMACR/P504S (U.S. Pat. No. 6,262,245); PCA3 (U.S. Pat. No. 7,008,765); PCGEMl (U.S. Pat. No. 6,828,429); prostein/P501 S, P503S, P504S, P509S, P510S, prostase/P703P, P710P (U.S. Publication No. 20030185830); RAS/KRAS (Bos, Cancer Res.
49:4682-89 (1989); Kranenburg, Biochimica et Biophysica Acta 1756:81-82 (2005)); and, those disclosed in U.S. Pat. Nos. 5,854,206 and 6,034,218, 7,229,774, each of which is herein incorporated by reference in its entirety. Markers for other cancers, diseases, infections, and metabolic conditions are also contemplated for inclusion in a multiplex or panel format.
In some embodiments, multiplex or array formats are utilized to detect multiple markers in combination. For example, in some embodiments, the level of expression of one or more, 2 or more, 3 or more, 4 or more, 5 or more, 10 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more non-coding RNAs (ncRNAs) is utilized in the research, screening, diagnostic and prognositic compositions and methods described herein. The one or more ncRNAs may be selected from the group comprising.
i. DNA and RNA Detection
The non-coding RNAs of the present disclosure are detected using a variety of nucleic acid techniques known to those of ordinary skill in the art, including but not limited to: nucleic acid sequencing; nucleic acid hybridization; and, nucleic acid amplification.
The methods, compositions and kits may comprise one or more ncRNAs. The methods, compositions and kits may comprise 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 15 or more, 20 or more, 25 or more, 30 or more, 40 or more, 45 or more, 50 or more, 55 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more, 130 or more, 140 or more, 150 or more ncRNAs.
The one or more ncRNAs may be selected from, for example, those described in SEQ ID
NOs: 1-2309.
1. Sequencing
In some embodiments, nucleic acid sequencing methods are utilized (e.g., for detection of amplified nucleic acids). In some embodiments, the technology provided herein finds use in a Second Generation (a.k.a. Next Generation or Next-Gen), Third Generation (a.k.a. Next-Next-Gen), or Fourth Generation (a.k.a. N3-Gen) sequencing technology including, but not limited to, pyrosequencing, sequencing-by-ligation, single molecule sequencing, sequence-by-synthesis (SBS), semiconductor sequencing, massive parallel clonal, massive parallel single molecule SBS, massive parallel single molecule real-time, massive parallel single molecule real-time nanopore technology, etc. Morozova and Marra provide a review of some such technologies in Genomics, 92: 255 (2008), herein incorporated by reference in its entirety. Those of ordinary skill in the art will recognize that because RNA is less stable in the cell and more prone to nuclease attack experimentally RNA is usually reverse transcribed to DNA before sequencing.
A number of DNA sequencing techniques are suitable, including fluorescence-based sequencing methodologies (See, e.g., Birren et al, Genome Analysis: Analyzing DNA, 1, Cold Spring Harbor, N.Y.; herein incorporated by reference in its entirety). In some embodiments, the technology finds use in automated sequencing techniques understood in that art. In some embodiments, the present technology finds use in parallel sequencing of partitioned amplicons (PCT Publication No:
WO2006084132 to Kevin McKernan et al, herein incorporated by reference in its entirety). In some embodiments, the technology finds use in DNA sequencing by parallel oligonucleotide extension
(See, e.g., U.S. Pat. No. 5,750,341 to Macevicz et al, and U.S. Pat. No. 6,306,597 to Macevicz et al., both of which are herein incorporated by reference in their entireties). Additional examples of sequencing techniques in which the technology finds use include the Church polony technology (Mitra et al., 2003, Analytical Biochemistry 320, 55-65; Shendure et al, 2005 Science 309, 1728-
1732; U.S. Pat. No. 6,432,360, U.S. Pat. No. 6,485,944, U.S. Pat. No. 6,511,803; herein incorporated by reference in their entireties), the 454 picotiter pyrosequencing technology (Margulies et al, 2005 Nature 437, 376-380; US 20050130173; herein incorporated by reference in their entireties), the Solexa single base addition technology (Bennett et al, 2005, Pharmacogenomics, 6, 373-382; U.S. Pat. No. 6,787,308; U.S. Pat. No. 6,833,246; herein incorporated by reference in their entireties), the Lynx massively parallel signature sequencing technology (Brenner et al. (2000). Nat. Biotechnol. 18:630-634; U.S. Pat. No. 5,695,934; U.S. Pat. No. 5,714,330; herein incorporated by reference in their entireties), and the Adessi PCR colony technology (Adessi et al. (2000). Nucleic Acid Res. 28, E87; WO 00018957; herein incorporated by reference in its entirety).
Next-generation sequencing (NGS) methods share the common feature of massively parallel, high-throughput strategies, with the goal of lower costs in comparison to older sequencing methods (see, e.g., Voelkerding et al., Clinical Chem., 55: 641-658, 2009; MacLean et al., Nature Rev.
Microbiol., 7: 287-296; each herein incorporated by reference in their entirety). NGS methods can be broadly divided into those that typically use template amplification and those that do not.
Amplification-requiring methods include pyrosequencing commercialized by Roche as the 454 technology platforms (e.g., GS 20 and GS FLX), Life Technologies/Ion Torrent, the Solexa platform commercialized by Illumina, GnuBio, and the Supported Oligonucleotide Ligation and Detection (SOLiD) platform commercialized by Applied Biosystems. Non-amplification approaches, also known as single-molecule sequencing, are exemplified by the HeliScope platform commercialized by Helicos Biosciences, and emerging platforms commercialized by VisiGen, Oxford Nanopore Technologies Ltd., and Pacific Biosciences, respectively.
In pyrosequencing (Voelkerding et al., Clinical Chem., 55: 641-658, 2009; MacLean et al., Nature Rev. Microbiol, 7: 287-296; U.S. Pat. No. 6,210,891 ; U.S. Pat. No. 6,258,568; each herein incorporated by reference in its entirety), template DNA is fragmented, end-repaired, ligated to adaptors, and clonally amplified in-situ by capturing single template molecules with beads bearing oligonucleotides complementary to the adaptors. Each bead bearing a single template type is compartmentalized into a water-in-oil microvesicle, and the template is clonally amplified using a technique referred to as emulsion PCR. The emulsion is disrupted after amplification and beads are deposited into individual wells of a picotitre plate functioning as a flow cell during the sequencing reactions. Ordered, iterative introduction of each of the four dNTP reagents occurs in the flow cell in the presence of sequencing enzymes and luminescent reporter such as luciferase. In the event that an appropriate dNTP is added to the 3' end of the sequencing primer, the resulting production of ATP causes a burst of luminescence within the well, which is recorded using a CCD camera. It is possible
to achieve read lengths greater than or equal to 400 bases, and 10 sequence reads can be achieved, resulting in up to 500 million base pairs (Mb) of sequence.
In the Solexa/Illumina platform (Voelkerding et al, Clinical Chem., 55: 641-658, 2009; MacLean et al, Nature Rev. Microbiol, 7: 287-296; U.S. Pat. No. 6,833,246; U.S. Pat. No.
7,115,400; U.S. Pat. No. 6,969,488; each herein incorporated by reference in its entirety), sequencing data are produced in the form of shorter-length reads. In this method, single-stranded fragmented DNA is end-repaired to generate 5'-phosphorylated blunt ends, followed by Klenow-mediated addition of a single A base to the 3' end of the fragments. A-addition facilitates addition of T- overhang adaptor oligonucleotides, which are subsequently used to capture the template-adaptor molecules on the surface of a flow cell that is studded with oligonucleotide anchors. The anchor is used as a PCR primer, but because of the length of the template and its proximity to other nearby anchor oligonucleotides, extension by PCR results in the "arching over" of the molecule to hybridize with an adjacent anchor oligonucleotide to form a bridge structure on the surface of the flow cell. These loops of DNA are denatured and cleaved. Forward strands are then sequenced with reversible dye terminators. The sequence of incorporated nucleotides is determined by detection of post- incorporation fluorescence, with each fluor and block removed prior to the next cycle of dNTP addition. Sequence read length ranges from 36 nucleotides to over 250 nucleotides, with overall output exceeding 1 billion nucleotide pairs per analytical run.
Sequencing nucleic acid molecules using SOLiD technology (Voelkerding et al, Clinical Chem., 55: 641-658, 2009; MacLean et al, Nature Rev. Microbiol, 7: 287-296; U.S. Pat. No.
5,912,148; U.S. Pat. No. 6,130,073; each herein incorporated by reference in their entirety) also involves fragmentation of the template, ligation to oligonucleotide adaptors, attachment to beads, and clonal amplification by emulsion PCR. Following this, beads bearing template are immobilized on a derivatized surface of a glass flow-cell, and a primer complementary to the adaptor oligonucleotide is annealed. However, rather than utilizing this primer for 3' extension, it is instead used to provide a 5' phosphate group for ligation to interrogation probes containing two probe-specific bases followed by 6 degenerate bases and one of four fluorescent labels. In the SOLiD system, interrogation probes have 16 possible combinations of the two bases at the 3' end of each probe, and one of four fluors at the 5' end. Fluor color, and thus identity of each probe, corresponds to specified color-space coding schemes. Multiple rounds (usually 7) of probe annealing, ligation, and fluor detection are followed by denaturation, and then a second round of sequencing using a primer that is offset by one base relative to the initial primer. In this manner, the template sequence can be computationally reconstructed, and template bases are interrogated twice, resulting in increased accuracy. Sequence read length averages 35 nucleotides, and overall output exceeds 4 billion bases per sequencing run.
In certain embodiments, the technology finds use in nanopore sequencing (see, e.g., Astier et al, J. Am. Chem. Soc. 2006 Feb 8; 128(5): 1705-10, herein incorporated by reference). The theory behind nanopore sequencing has to do with what occurs when a nanopore is immersed in a conducting fluid and a potential (voltage) is applied across it. Under these conditions a slight electric current due to conduction of ions through the nanopore can be observed, and the amount of current is exceedingly sensitive to the size of the nanopore. As each base of a nucleic acid passes through the nanopore, this causes a change in the magnitude of the current through the nanopore that is distinct for each of the four bases, thereby allowing the sequence of the DNA molecule to be determined.
In certain embodiments, the technology finds use in HeliScope by Helicos Biosciences (Voelkerding et al., Clinical Chem., 55: 641-658, 2009; MacLean et al., Nature Rev. Microbiol., 7: 287-296; U.S. Pat. No. 7,169,560; U.S. Pat. No. 7,282,337; U.S. Pat. No. 7,482,120; U.S. Pat. No. 7,501,245; U.S. Pat. No. 6,818,395; U.S. Pat. No. 6,911,345; U.S. Pat. No. 7,501,245; each herein incorporated by reference in their entirety). Template DNA is fragmented and polyadenylated at the 3' end, with the final adenosine bearing a fluorescent label. Denatured polyadenylated template fragments are ligated to poly(dT) oligonucleotides on the surface of a flow cell. Initial physical locations of captured template molecules are recorded by a CCD camera, and then label is cleaved and washed away. Sequencing is achieved by addition of polymerase and serial addition of fluorescently -labeled dNTP reagents. Incorporation events result in fluor signal corresponding to the dNTP, and signal is captured by a CCD camera before each round of dNTP addition. Sequence read length ranges from 25-50 nucleotides, with overall output exceeding 1 billion nucleotide pairs per analytical run.
The Ion Torrent technology is a method of DNA sequencing based on the detection of hydrogen ions that are released during the polymerization of DNA (see, e.g., Science 327(5970): 1190 (2010); U.S. Pat. Appl. Pub. Nos. 20090026082, 20090127589, 20100301398, 20100197507, 20100188073, and 20100137143, incorporated by reference in their entireties for all purposes). A microwell contains a template DNA strand to be sequenced. Beneath the layer of microwells is a hypersensitive ISFET ion sensor. All layers are contained within a CMOS semiconductor chip, similar to that used in the electronics industry. When a dNTP is incorporated into the growing complementary strand a hydrogen ion is released, which triggers a hypersensitive ion sensor. If homopolymer repeats are present in the template sequence, multiple dNTP molecules will be incorporated in a single cycle. This leads to a corresponding number of released hydrogens and a proportionally higher electronic signal. This technology differs from other sequencing technologies in that no modified nucleotides or optics are used. The per-base accuracy of the Ion Torrent sequencer is -99.6% for 50 base reads, with -100 Mb to 100Gb generated per run. The read-length is
100-300 base pairs. The accuracy for homopolymer repeats of 5 repeats in length is -98%. The benefits of ion semiconductor sequencing are rapid sequencing speed and low upfront and operating costs.
The technology finds use in another nucleic acid sequencing approach developed by Stratos Genomics, Inc. and involves the use of Xpandomers. This sequencing process typically includes providing a daughter strand produced by a template-directed synthesis. The daughter strand generally includes a plurality of subunits coupled in a sequence corresponding to a contiguous nucleotide sequence of all or a portion of a target nucleic acid in which the individual subunits comprise a tether, at least one probe or nucleobase residue, and at least one selectively cleavable bond. The selectively cleavable bond(s) is/are cleaved to yield an Xpandomer of a length longer than the plurality of the subunits of the daughter strand. The Xpandomer typically includes the tethers and reporter elements for parsing genetic information in a sequence corresponding to the contiguous nucleotide sequence of all or a portion of the target nucleic acid. Reporter elements of the
Xpandomer are then detected. Additional details relating to Xpandomer-based approaches are described in, for example, U.S. Pat. Pub No. 20090035777, entitled "High Throughput Nucleic Acid Sequencing by Expansion," filed June 19, 2008, which is incorporated herein in its entirety.
Other emerging single molecule sequencing methods include real-time sequencing by synthesis using a VisiGen platform (Voelkerding et al., Clinical Chem., 55: 641-58, 2009; U.S. Pat. No. 7,329,492; U.S. Pat. App. Ser. No. 11/671956; U.S. Pat. App. Ser. No. 11/781166; each herein incorporated by reference in their entirety) in which immobilized, primed DNA template is subjected to strand extension using a fluorescently-modified polymerase and florescent acceptor molecules, resulting in detectible fluorescence resonance energy transfer (FRET) upon nucleotide addition.
2. Hybridization
Illustrative non-limiting examples of nucleic acid hybridization techniques include, but are not limited to, in situ hybridization (ISH), microarray, and Southern or Northern blot.
In situ hybridization (ISH) is a type of hybridization that uses a labeled complementary DNA or RNA strand as a probe to localize a specific DNA or RNA sequence in a portion or section of tissue (in situ), or, if the tissue is small enough, the entire tissue (whole mount ISH). DNA ISH can be used to determine the structure of chromosomes. RNA ISH is used to measure and localize mRNAs and other transcripts (e.g., ncRNAs) within tissue sections or whole mounts. Sample cells and tissues are usually treated to fix the target transcripts in place and to increase access of the probe. The probe hybridizes to the target sequence at elevated temperature, and then the excess probe is washed away. The probe that was labeled with either radio-, fluorescent- or antigen-labeled bases is localized and quantitated in the tissue using either autoradiography, fluorescence microscopy or
immunohistochemistry, respectively. ISH can also use two or more probes, labeled with radioactivity or the other non-radioactive labels, to simultaneously detect two or more transcripts.
In some embodiments, ncRNAs are detected using fluorescence in situ hybridization (FISH). In some embodiments, FISH assays utilize bacterial artificial chromosomes (BACs). These have been used extensively in the human genome sequencing project (see Nature 409: 953-958 (2001)) and clones containing specific BACs are available through distributors that can be located through many sources, e.g., NCBI. Each BAC clone from the human genome has been given a reference name that unambiguously identifies it. These names can be used to find a corresponding GenBank sequence and to order copies of the clone from a distributor.
The present disclosure further provides a method of performing a FISH assay on the patient sample. The methods disclosed herein may comprise performing a FISH assay on one or more cells, tissues, organs, or fluids surrounding such cells, tissues and organs. In some instances, the methods disclosed herein further comprise performing a FISH assy on human prostate cells, human prostate tissue or on the fluid surrounding said human prostate cells or human prostate tissue. Alternatively, or additionally, the methods disclosed herien comprise performing a FISH assay on breast cells, lung cells, pancreatic cells, liver cells, breast tissue, lung tissue, pancreatic tissue, liver tissue, or on the fluid surrounding the cells or tissues. Specific protocols are well known in the art and can be readily adapted for the present disclosure. Guidance regarding methodology may be obtained from many references including: In situ Hybridization: Medical Applications (eds. G. R. Coulton and J. de Belleroche), Kluwer Academic Publishers, Boston (1992); In situ Hybridization: In Neurobiology; Advances in Methodology (eds. J. H. Eberwine, K. L. Valentino, and J. D. Barchas), Oxford University Press Inc., England (1994); In situ Hybridization: A Practical Approach (ed. D. G.
Wilkinson), Oxford University Press Inc., England (1992)); Kuo, et al. , Am. J. Hum. Genet. 49. W2- 119 (1991); Klinger, et al. , Am. J. Hum. Genet. 57:55-65 (1992); and Ward, et al. , Am. J. Hum. Genet. 52:854-865 (1993)). There are also kits that are commercially available and that provide protocols for performing FISH assays (available from e.g. , Oncor, Inc., Gaithersburg, MD). Patents providing guidance on methodology include U.S. 5,225,326; 5,545,524; 6,121,489 and 6,573,043. All of these references are hereby incorporated by reference in their entirety and may be used along with similar references in the art and with the information provided in the Examples section herein to establish procedural steps convenient for a particular laboratory.
The one or more ncRNAs may be detected by conducting one or more hybridization reactions. The one or more hybridization reactions may comprise one or more hybridization arrays, hybridization reactions, hybridization chain reactions, isothermal hybridization reactions, nucleic acid hybridization reactions, or a combination thereof. The one or more hybridization arrays may
comprise hybridization array genotyping, hybridization array proportional sensing, DNA hybridization arrays, macroarrays, microarrays, high-density oligonucleotide arrays, genomic hybridization arrays, comparative hybridization arrays, or a combination thereof.
3. Microarrays
Different kinds of biological assays are called microarrays including, but not limited to:
DNA microarrays (e.g. , cDNA microarrays and oligonucleotide microarrays); protein microarrays; tissue microarrays; transfection or cell microarrays; chemical compound microarrays; and, antibody microarrays. A DNA microarray, commonly known as gene chip, DNA chip, or biochip, is a collection of microscopic DNA spots attached to a solid surface (e.g. , glass, plastic or silicon chip) forming an array for the purpose of expression profiling or monitoring expression levels for thousands of genes simultaneously. The affixed DNA segments are known as probes, thousands of which can be used in a single DNA microarray. Microarrays can be used to identify disease genes or transcripts (e.g., ncRNAs) by comparing gene expression in disease and normal cells. Microarrays can be fabricated using a variety of technologies, including but not limiting: printing with fine- pointed pins onto glass slides; photolithography using pre-made masks; photolithography using dynamic micromirror devices; ink-jet printing; or, electrochemistry on microelectrode arrays.
3. Amplification
The methods disclosed herein may comprise conducting one or more amplification reactions. Nucleic acids (e.g., ncRNAs) may be amplified prior to or simultaneous with detection. Conducting one or more amplification reactions may comprise one or more PCR-based amplifications, non-PCR based amplifications, or a combination thereof. Illustrative non-limiting examples of nucleic acid amplification techniques include, but are not limited to, polymerase chain reaction (PCR), reverse transcription polymerase chain reaction (RT-PCR), nested PCR, linear amplification, multiple displacement amplification (MDA), real-time SDA, rolling circle amplification, circle-to-circle amplification transcription-mediated amplification (TMA), ligase chain reaction (LCR), strand displacement amplification (SDA), and nucleic acid sequence based amplification (NASBA). Those of ordinary skill in the art will recognize that certain amplification techniques (e.g. , PCR) require that RNA be reversed transcribed to DNA prior to amplification (e.g. , RT-PCR), whereas other amplification techniques directly amplify RNA (e.g., TMA and NASBA).
The polymerase chain reaction (U.S. Pat. Nos. 4,683,195, 4,683,202, 4,800,159 and
4,965,188, each of which is herein incorporated by reference in its entirety), commonly referred to as PCR, uses multiple cycles of denaturation, annealing of primer pairs to opposite strands, and primer extension to exponentially increase copy numbers of a target nucleic acid sequence. In a variation called RT-PCR, reverse transcriptase (RT) is used to make a complementary DNA (cDNA) from
mRNA, and the cDNA is then amplified by PCR to produce multiple copies of DNA. For other various permutations of PCR see, e.g. , U.S. Pat. Nos. 4,683,195, 4,683,202 and 4,800,159; Mullis et al, Meth. Enzymol. 155: 335 (1987); and, Murakawa et al, DNA 7: 287 (1988), each of which is herein incorporated by reference in its entirety.
Transcription mediated amplification (U.S. Pat. Nos. 5,480,784 and 5,399,491, each of which is herein incorporated by reference in its entirety), commonly referred to as TMA, synthesizes multiple copies of a target nucleic acid sequence autocatalytically under conditions of substantially constant temperature, ionic strength, and pH in which multiple RNA copies of the target sequence autocatalytically generate additional copies. See, e.g. , U.S. Pat. Nos. 5,399,491 and 5,824,518, each of which is herein incorporated by reference in its entirety. In a variation described in U.S. Publ. No. 20060046265 (herein incorporated by reference in its entirety), TMA optionally incorporates the use of blocking moieties, terminating moieties, and other modifying moieties to improve TMA process sensitivity and accuracy.
The ligase chain reaction (Weiss, R., Science 254: 1292 (1991), herein incorporated by reference in its entirety), commonly referred to as LCR, uses two sets of complementary DNA oligonucleotides that hybridize to adjacent regions of the target nucleic acid. The DNA
oligonucleotides are covalently linked by a DNA ligase in repeated cycles of thermal denaturation, hybridization and ligation to produce a detectable double-stranded ligated oligonucleotide product.
Strand displacement amplification (Walker, G. et al, Proc. Natl. Acad. Sci. USA 89: 392-396 (1992); U.S. Pat. Nos. 5,270,184 and 5,455,166, each of which is herein incorporated by reference in its entirety), commonly referred to as SDA, uses cycles of annealing pairs of primer sequences to opposite strands of a target sequence, primer extension in the presence of a dNTPaS to produce a duplex hemiphosphorothioated primer extension product, endonuclease-mediated nicking of a hemimodified restriction endonuclease recognition site, and polymerase-mediated primer extension from the 3' end of the nick to displace an existing strand and produce a strand for the next round of primer annealing, nicking and strand displacement, resulting in geometric amplification of product. Thermophilic SDA (tSDA) uses thermophilic endonucleases and polymerases at higher temperatures in essentially the same method (EP Pat. No. 0 684 315).
Other amplification methods include, for example: nucleic acid sequence based amplification (U.S. Pat. No. 5,130,238, herein incorporated by reference in its entirety), commonly referred to as NASBA; one that uses an RNA replicase to amplify the probe molecule itself (Lizardi et al, BioTechnol. 6: 1197 (1988), herein incorporated by reference in its entirety), commonly referred to as replicase; a transcription based amplification method (Kwoh et al, Proc. Natl. Acad. Sci. USA 86: 1173 (1989)); and, self-sustained sequence replication (Guatelli et al, Proc. Natl. Acad. Sci. USA
87: 1874 (1990), each of which is herein incorporated by reference in its entirety). For further discussion of known amplification methods see Persing, David H., "In Vitro Nucleic Acid
Amplification Techniques" in Diagnostic Medical Microbiology: Principles and Applications (Persing et al., Eds.), pp. 51-87 (American Society for Microbiology, Washington, DC (1993)). ii. Data Analysis
In some embodiments, a computer-based analysis program is used to translate the raw data generated by the detection assay (e.g. , the presence, absence, or amount of a given marker or markers) into data of predictive value for a clinician. The clinician can access the predictive data using any suitable means. Thus, in some preferred embodiments, the present disclosure provides the further benefit that the clinician, who is not likely to be trained in genetics or molecular biology, need not understand the raw data. The data is presented directly to the clinician in its most useful form. The clinician is then able to immediately utilize the information in order to optimize the care of the subject.
The present disclosure contemplates any method capable of receiving, processing, and transmitting the information to and from laboratories conducting the assays, information providers, medical personnel, and subjects. For example, in some embodiments of the present disclosure, a sample (e.g., a biopsy or a serum or urine sample) is obtained from a subject and submitted to a profiling service (e.g., clinical lab at a medical facility, genomic profiling business, etc.), located in any part of the world (e.g. , in a country different than the country where the subject resides or where the information is ultimately used) to generate raw data. Where the sample comprises a tissue or other biological sample, the subject may visit a medical center to have the sample obtained and sent to the profiling center, or subjects may collect the sample themselves (e.g. , a urine sample) and directly send it to a profiling center. Where the sample comprises previously determined biological information, the information may be directly sent to the profiling service by the subject (e.g. , an information card containing the information may be scanned by a computer and the data transmitted to a computer of the profiling center using an electronic communication systems). Once received by the profiling service, the sample is processed and a profile is produced (i.e. , expression data), specific for the diagnostic or prognostic information desired for the subject.
The profile data is then prepared in a format suitable for interpretation by one or more medical personnel (e.g., a treating clinician, physician assistant, nurse, or pharmacist). For example, rather than providing raw expression data, the prepared format may represent a diagnosis or risk assessment (e.g. , presence or absence of a ncRNA) for the subject, along with recommendations for particular treatment options. The data may be displayed to the medical personnel by any suitable method. For example, in some embodiments, the profiling service generates a report that can be
printed for the medical personnel (e.g. , at the point of care) or displayed to the medical personnel on a computer monitor.
In some embodiments, the information is first analyzed at the point of care or at a regional facility. The raw data is then sent to a central processing facility for further analysis and/or to convert the raw data to information useful for medical personnel or patient. The central processing facility provides the advantage of privacy (all data is stored in a central facility with uniform security protocols), speed, and uniformity of data analysis. The central processing facility can then control the fate of the data following treatment of the subject. For example, using an electronic communication system, the central facility can provide data to the medical personnel, the subject, or researchers.
In some embodiments, the subject is able to directly access the data using the electronic communication system. The subject may chose further intervention or counseling based on the results.
In some embodiments, the data is used for research use. For example, the data may be used to further optimize the inclusion or elimination of markers as useful indicators of a particular condition or stage of disease or as a companion diagnostic to determine a treatment course of action,
iii. Compositions & Kits
Compositions for use in the diagnostic methods described herein include, but are not limited to, probes, amplification oligonucleotides, and the like.
The compositions and kits may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more,
14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more probes.
The probes may hybridize to 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more,
15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more target molecules. The target molecules may be a ncRNA, RNA, DNA, cDNA, mRNA, a portion or fragment thereof or a combination thereof. In some instances, at least a portion of the target molecules are ncRNAs. The probes may hybridize to 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more ncRNAs disclosed herein (e.g., SEQ ID NOs: 1-2309).
Typically, the probes comprise a target specific sequence. The target specific sequence may be complementary to at least a portion of the target molecule. The target specific sequence may be at least about 50% or more, 55% or more, 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 90% or more, 95% or more, 97% or more, 98% or more, or 100% complementary to at leat a portion of the target molecule.
The target specific sequence may be at least about 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 16 or more, 17 or more, 18 or more, 19 or more, 20 or more nucleotides in length. In some instances, the target specific sequence is between about 8 to about 20 nucleotides, 10 to about 18 nucleotides, or 12 to about 16 nucleotides in length.
The compositions and kits may comprise a plurality of probes, wherein the two or more probes of the plurality of probes comprise identical target specific sequences. The compositions and kits may comprise a plurality of probes, wherein the two or more probes of the plurality of probes comprise different target specific sequences.
The probes may further comprise a unique sequence. The unique sequence is
noncomplementary to the ncRNA. The unique sequence may comprise a label, barcode, or unique identifier. The unique sequence may comprise a random sequence, nonrandom sequence, or a combination thereof. The unique sequence may be at least about 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 16 or more, 17 or more, 18 or more, 19 or more, 20 or more, 22 or more, 24 or more, 26 or more, 28 or more, 30 or more nucleotides in length. In some instances, the unique sequence is between about 8 to about 20 nucleotides, 10 to about 18 nucleotides, or 12 to about 16 nucleotides in length.
The unique sequence may allow differentiation of two or more target molecules. The two or more target molecules may have identical sequences. Thus, the unique sequence may allow quantification of a target molecule. Alternatively, the two or more target molecules may have different sequences. Thus, the unique sequence may allow detection of the target molecules. The compositions and kits may comprise a plurality of probes for quantifying one or more target molecules. The compositions and kits may comprise a plurality of probes for detecting one or more target molecules.
The unique sequence may allow differentiation of two or more samples. The compositions and kits may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more probe sets for differentiating two or more samples from one or more subjects. The two or more samples may be from two or more different subjects. For example, the
compositions and kits comprise a first set of probes comprising a first unique sequence that is specific for a first subject and a second set of probes comprosing a second unique sequence that is specific for a second subject. The compositions and kits may further comprise one or more sets of probes with one or more unique sequences to differentiate one or more additional subjects.
The compositions and kits may comprise 2 or more probe sets for differentiating from 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more samples from 1 or more subjects.
The compositions and kits may comprise 2 or more probe sets for differentiating 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more samples from one or more cells, tissues, organs, bodily fluid, or a combination thereof.
The compositions and kits may comprise 2 or more probe sets for differentiating samples from 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more subjects.
Alternatively, or additionally, the two or more samples may be from two or more different timepoints from the same subject or different subjects. For example, the compositions and kits comprise a first set of probes comprising a first unique sequence that is specific for a first subject and a second set of probes comprosing a second unique sequence that is specific for a second subject. The compositions and kits may comprise 2 or more probe sets for differentiating samples from 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more timepoints. The timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more hours. The timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more days. The timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more weeks. The timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more months. The timepoints may be every 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 or more years. The timepoints may be before diagnosis, after diagnosis, before treatment, during treatment, after treatment, before metastasis, after metastatis, before remission, during remission, or a combination thereof.
The compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific
sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are identical and the first unique sequence and the second unique sequence are different. The compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are different and the first unique sequence and the second unique sequence are different. The compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are identical and the first unique sequence and the second unique sequence are identical. The compositions and kits may comprise a first probe comprising a first target-specific sequence and a first unique sequence and a second probe comprising a second target-specific sequence and a second unique sequence, wherein the first target specific sequence and the second target specific sequence are different and the first unique sequence and the second unique sequence are identical.
The probes may further comprise a universal sequence. The universal sequence may comprise a primer binding site. The universal sequence may enable detection of the target sequence. The universal sequence may enable amplification of the target sequence. The universal sequence may enable transcription or reverse transcription of the target sequence. The universal sequence may enable sequencing of the target sequence.
The probe and antibody compositions of the present disclosure may also be provided on a solid support. The solid support may comprise one or more beads, plates, solid surfaces, wells, chips, or a combination thereof. The beads may be magnetic, antibody coated, protein A crosslinked, protein G crosslinked, streptavidin coated, oligonucleotide conjugated, silica coated, or a combination thereof. Examples of beads include, but are not limited to, Ampure beads, AMPure XP beads, streptavidin beads, agarose beads, magnetic beads, Dynabeads®, MACS® microbeads, antibody conjugated beads (e.g., anti-immunoglobulin microbead), protein A conjugated beads, protein G conjugated beads, protein A/G conjugated beads, protein L conjugated beads, oligo-dT conjugated beads, silica beads, silica-like beads, anti-biotin microbead, anti-fluorochrome microbead, and BcMag™ Carboxy-Terminated Magnetic Beads.
The compositions and kits may comprise primers and primer pairs capable of amplifying target molecules, or fragments or subsequences or complements thereof. The nucleotide sequences of the target molecules may be provided in computer-readable media for in silico applications and as a basis for the design of appropriate primers for amplification of one or more target molecules.
Primers based on the nucleotide sequences of target molecules can be designed for use in amplification of the target molecules. For use in amplification reactions such as PCR, a pair of primers can be used. The exact composition of the primer sequences is not critical to the disclosure, but for most applications the primers may hybridize to specific sequences of the target molecules or the universal sequence of the probe under stringent conditions, particularly under conditions of high stringency, as known in the art. The pairs of primers are usually chosen so as to generate an amplification product of at least about 15 or more, 20 or more, 30 or more, 40 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 125 or more, 150 or more, 175 or more, 200 or more, 250 or more, 300 or more, 350 or more, 400 or more, 450 or more, 500 or more, 600 or more, 700 or more, 800 or more, 900 or more, or 1000 or more nucleotides. Algorithms for the selection of primer sequences are generally known, and are available in commercial software packages. These primers may be used in standard quantitative or qualitative PCR-based assays to assess transcript expression levels of target molecules. Alternatively, these primers may be used in combination with probes, such as molecular beacons in amplifications using real-time PCR.
One skilled in the art also appreciates that the nucleotide sequence of the entire length of the primer does not need to be derived from the target sequence. Thus, for example, the primer may comprise nucleotide sequences at the 5' and/or 3' termini that are not derived from the target molecule. Nucleotide sequences which are not derived from the nucleotide sequence of the target molecule may provide additional functionality to the primer. For example, they may provide a restriction enzyme recognition sequence or a "tag" that facilitates detection, isolation, purification or immobilization onto a solid support. Alternatively, the additional nucleotides may provide a self- complementary sequence that allows the primer to adopt a hairpin configuration. Such configurations may be necessary for certain primers, for example, molecular beacon and Scorpion primers, which can be used in solution hybridization techniques.
The probes or primers can incorporate moieties useful in detection, isolation, purification, or immobilization, if desired. Such moieties are well-known in the art (see, for example, Ausubel et al, (1997 & updates) Current Protocols in Molecular Biology, Wiley & Sons, New York) and are chosen such that the ability of the probe to hybridize with its target molecule is not affected.
Examples of suitable moieties are detectable labels, such as radioisotopes, fluorophores, chemiluminophores, enzymes, colloidal particles, and fluorescent microparticles, as well as antigens, antibodies, haptens, avidin/streptavidin, biotin, haptens, enzyme cofactors / substrates, enzymes, and the like.
A label can optionally be attached to or incorporated into a probe or primer to allow detection and/or quantitation of a target polynucleotide representing the target molecule of interest. The target
polynucleotide may be the expressed target molecule RNA itself, a cDNA copy thereof, or an amplification product derived therefrom, and may be the positive or negative strand, so long as it can be specifically detected in the assay being used. Similarly, an antibody may be labeled.
In certain multiplex formats, labels used for detecting different target molecules may be distinguishable. The label can be attached directly (e.g., via covalent linkage) or indirectly, e.g., via a bridging molecule or series of molecules (e.g., a molecule or complex that can bind to an assay component, or via members of a binding pair that can be incorporated into assay components, e.g. biotin-avidin or streptavidin). Many labels are commercially available in activated forms which can readily be used for such conjugation (for example through amine acylation), or labels may be attached through known or determinable conjugation schemes, many of which are known in the art.
Labels useful in the disclosure described herein include any substance which can be detected when bound to or incorporated into the target molecule. Any effective detection method can be used, including optical, spectroscopic, electrical, piezoelectrical, magnetic, Raman scattering, surface plasmon resonance, colorimetric, calorimetric, etc. A label is typically selected from a chromophore, a lumiphore, a fluorophore, one member of a quenching system, a chromogen, a hapten, an antigen, a magnetic particle, a material exhibiting nonlinear optics, a semiconductor nanocrystal, a metal nanoparticle, an enzyme, an antibody or binding portion or equivalent thereof, an aptamer, and one member of a binding pair, and combinations thereof. Quenching schemes may be used, wherein a quencher and a fluorophore as members of a quenching pair may be used on a probe, such that a change in optical parameters occurs upon binding to the target introduce or quench the signal from the fluorophore. One example of such a system is a molecular beacon. Suitable quencher/fluorophore systems are known in the art. The label may be bound through a variety of intermediate linkages. For example, a target polynucleotide may comprise a biotin-binding species, and an optically detectable label may be conjugated to biotin and then bound to the labeled target polynucleotide. Similarly, a polynucleotide sensor may comprise an immunological species such as an antibody or fragment, and a secondary antibody containing an optically detectable label may be added.
Chromophores useful in the methods described herein include any substance which can absorb energy and emit light. For multiplexed assays, a plurality of different signaling chromophores can be used with detectably different emission spectra. The chromophore can be a lumophore or a fluorophore. Typical fluorophores include fluorescent dyes, semiconductor nanocrystals, lanthanide chelates, polynucleotide-specific dyes and green fluorescent protein.
Coding schemes may optionally be used, comprising encoded particles and/or encoded tags associated with different polynucleotides of the disclosure. A variety of different coding schemes are known in the art, including fluorophores, including SCNCs, deposited metals, and RF tags.
Polynucleotides from the described target molecules may be employed as probes for detecting target molecules expression, for ligation amplification schemes, or may be used as primers for amplification schemes of all or a portion of a target molecule. When amplified, either strand produced by amplification may be provided in purified and/or isolated form.
In some instances, the compositions and kits comprise a biomarker library. The biomarker library may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more target molecules. The target molecules may be a ncRNA, RNA, DNA, cDNA, mRNA, a portion or fragment thereof or a combination thereof. In some instances, at least a portion of the target molecules are ncRNAs. The biomarker library may comprise 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 11 or more, 12 or more, 13 or more, 14 or more, 15 or more, 20 or more, 25 or more, 30 or more, 35 or more, 40 or more, 45 or more, 50 or more, 60 or more, 70 or more, 80 or more, 90 or more, 100 or more, 110 or more, 120 or more ncRNAs disclosed herein.
In some embodiments, is a kit for analyzing a cancer comprising (a) a probe set comprising a plurality of probes comprising target specific sequences complementary to one or more target molecules, wherein the one or more target molecules comprise one or more ncRNAs; and (b) a computer model or algorithm for analyzing an expression level and/or expression profile of the one or more target molecules in a sample. The target molecules may comprose one or more of those described by SEQ ID NOs: 1 -2309, or a combination thereof.
In some embodiments, is a kit for analyzing a cancer comprising (a) a probe set comprising a plurality of probes comprising target specific sequences complementary to one or more target molecules of a biomarker library; and (b) a computer model or algorithm for analyzing an expression level and/or expression profile of the one or more target molecules in a sample. Control samples and/or nucleic acids may optionally be provided in the kit. Control samples may include tissue and/or nucleic acids obtained from or representative of tumor samples from a healthy subject, as well as tissue and/or nucleic acids obtained from or representative of tumor samples from subjects diagnosed with a cancer.
Instructions for using the kit to perform one or more methods of the disclosure can be provided, and can be provided in any fixed medium. The instructions may be located inside or outside a container or housing, and/or may be printed on the interior or exterior of any surface
thereof. A kit may be in multiplex form for concurrently detecting and/or quantitating one or more different target polynucleotides representing the expressed target molecules,
iv. Devices
Devices useful for performing methods of the disclosure are also provided. The devices can comprise means for characterizing the expression level of a target molecule of the disclosure, for example components for performing one or more methods of nucleic acid extraction, amplification, and/or detection. Such components may include one or more of an amplification chamber (for example a thermal cycler), a plate reader, a spectrophotometer, capillary electrophoresis apparatus, a chip reader, and or robotic sample handling components. These components ultimately can obtain data that reflects the expression level of the target molecules used in the assay being employed.
The devices may include an excitation and/or a detection means. Any instrument that provides a wavelength that can excite a species of interest and is shorter than the emission wavelength(s) to be detected can be used for excitation. Commercially available devices can provide suitable excitation wavelengths as well as suitable detection component.
Exemplary excitation sources include a broadband UV light source such as a deuterium lamp with an appropriate filter, the output of a white light source such as a xenon lamp or a deuterium lamp after passing through a monochromator to extract out the desired wavelength(s), a continuous wave (cw) gas laser, a solid state diode laser, or any of the pulsed lasers. Emitted light can be detected through any suitable device or technique; many suitable approaches are known in the art. For example, a fluorimeter or spectrophotometer may be used to detect whether the test sample emits light of a wavelength characteristic of a label used in an assay.
The devices typically comprise a means for identifying a given sample, and of linking the results obtained to that sample. Such means can include manual labels, barcodes, and other indicators which can be linked to a sample vessel, and/or may optionally be included in the sample itself, for example where an encoded particle is added to the sample. The results may be linked to the sample, for example in a computer memory that contains a sample designation and a record of expression levels obtained from the sample. Linkage of the results to the sample can also include a linkage to a particular sample receptacle in the device, which is also linked to the sample identity.
The devices also comprise a means for correlating the expression levels of the target molecules being studied with a prognosis of disease outcome. Such means may comprise one or more of a variety of correlative techniques, including lookup tables, algorithms, multivariate models, and linear or nonlinear combinations of expression models or algorithms. The expression levels may be converted to one or more likelihood scores, reflecting a likelihood that the patient providing the sample may exhibit a particular disease outcome. The models and/or algorithms can be provided in
machine readable format and can optionally further designate a treatment modality for a patient or class of patients.
The device also comprises output means for outputting the disease status, prognosis and/or a treatment modality. Such output means can take any form which transmits the results to a patient and/or a healthcare provider, and may include a monitor, a printed format, or both. The device may use a computer system for performing one or more of the steps provided.
The methods disclosed herein may also comprise the transmission of data/information. For example, data/information derived from the detection and/or quantification of the target may be transmitted to another device and/or instrument. In some instances, the information obtained from an algorithm may also be transmitted to another device and/or instrument. Transmission of the data/information may comprise the transfer of data/information from a first source to a second source. The first and second sources may be in the same approximate location (e.g., within the same room, building, block, campus). Alternatively, first and second sources may be in multiple locations (e.g., multiple cities, states, countries, continents, etc).
Transmission of the data/information may comprise digital transmission or analog transmission. Digital transmission may comprise the physical transfer of data (a digital bit stream) over a point-to-point or point-to-multipoint communication channel. Examples of such channels are copper wires, optical fibres, wireless communication channels, and storage media. The data may be represented as an electromagnetic signal, such as an electrical voltage, radiowave, microwave, or infrared signal.
Analog transmission may comprise the transfer of a continuously varying analog signal. The messages can either be represented by a sequence of pulses by means of a line code (baseband transmission), or by a limited set of continuously varying wave forms (passband transmission), using a digital modulation method. The passband modulation and corresponding demodulation (also known as detection) can be carried out by modem equipment. According to the most common definition of digital signal, both baseband and passband signals representing bit-streams are considered as digital transmission, while an alternative definition only considers the baseband signal as digital, and passband transmission of digital data as a form of digital-to-analog conversion,
v. Samples
Samples for use with the compositions and kits and in the methods of the present disclosure comprise nucleic acids suitable for providing RNA expression information. In principle, the biological sample from which the expressed RNA is obtained and analyzed for target molecule expression can be any material suspected of comprising cancer tissue or cells. The sample can be a
biological sample used directly in a method of the disclosure. Alternatively, the sample can be a sample prepared from a biological sample.
In one embodiment, the sample or portion of the sample comprising or suspected of comprising cancer tissue or cells can be any source of biological material, including cells, tissue, secretions, or fluid, including bodily fluids. Non-limiting examples of the source of the sample include an aspirate, a needle biopsy, a cytology pellet, a bulk tissue preparation or a section thereof obtained for example by surgery or autopsy, lymph fluid, blood, plasma, serum, tumors, and organs. Alternatively, or additionally, the source of the sample can be urine, bile, excrement, sweat, tears, vaginal fluids, spinal fluid, and stool. In some instances, the sources of the sample are secretions. In some instances, the secretions are exosomes.
The samples may be archival samples, having a known and documented medical outcome, or may be samples from current patients whose ultimate medical outcome is not yet known.
In some embodiments, the sample may be dissected prior to molecular analysis. The sample may be prepared via macrodissection of a bulk tumor specimen or portion thereof, or may be treated via microdissection, for example via Laser Capture Microdissection (LCM).
The sample may initially be provided in a variety of states, as fresh tissue, fresh frozen tissue, fine needle aspirates, and may be fixed or unfixed. Frequently, medical laboratories routinely prepare medical samples in a fixed state, which facilitates tissue storage. A variety of fixatives can be used to fix tissue to stabilize the morphology of cells, and may be used alone or in combination with other agents. Exemplary fixatives include crosslinking agents, alcohols, acetone, Bouin's solution, Zenker solution, Hely solution, osmic acid solution and Camoy solution.
Crosslinking fixatives can comprise any agent suitable for forming two or more covalent bonds, for example, an aldehyde. Sources of aldehydes typically used for fixation include formaldehyde, paraformaldehyde, glutaraldehyde or formalin. Preferably, the crosslinking agent comprises formaldehyde, which may be included in its native form or in the form of
paraformaldehyde or formalin. One of skill in the art would appreciate that for samples in which crosslinking fixatives have been used special preparatory steps may be necessary including for example heating steps and proteinase-k digestion.
One or more alcohols may be used to fix tissue, alone or in combination with other fixatives. Exemplary alcohols used for fixation include methanol, ethanol and isopropanol.
Formalin fixation is frequently used in medical laboratories. Formalin comprises both an alcohol, typically methanol, and formaldehyde, both of which can act to fix a biological sample.
Whether fixed or unfixed, the biological sample may optionally be embedded in an embedding medium. Exemplary embedding media used in histology including paraffin, Tissue-Tek®
V.I.P.TM, Paramat, Paramat Extra, Paraplast, Paraplast X-tra, Paraplast Plus, Peel Away Paraffin Embedding Wax, Polyester Wax, Carbowax Polyethylene Glycol, PolyfinTM, Tissue Freezing Medium TFMFM, Cryo-GefTM, and OCT Compound (Electron Microscopy Sciences, Hatfield, PA). Prior to molecular analysis, the embedding material may be removed via any suitable techniques, as known in the art. For example, where the sample is embedded in wax, the embedding material may be removed by extraction with organic solvent(s), for example xylenes. Kits are commercially available for removing embedding media from tissues. Samples or sections thereof may be subjected to further processing steps as needed, for example serial hydration or dehydration steps.
In some embodiments, the sample is a fixed, wax-embedded biological sample. Frequently, samples from medical laboratories are provided as fixed, wax-embedded samples, most commonly as formalin-fixed, paraffin embedded (FFPE) tissues.
Whatever the source of the biological sample, the target polynucleotide that is ultimately assayed can be prepared synthetically (in the case of control sequences), but typically is purified from the biological source and subjected to one or more preparative steps. The RNA may be purified to remove or diminish one or more undesired components from the biological sample or to concentrate it. Conversely, where the RNA is too concentrated for the particular assay, it may be diluted.
II. Drug Screening Applications
In some embodiments, the present disclosure provides drug screening assays (e.g. , to screen for anticancer drugs). The screening methods of the present disclosure utilize ncRNAs. For example, in some embodiments, the present disclosure provides methods of screening for compounds that alter the expression or activity of ncRNAs. The compounds may increase the expression or activity of the ncRNAs. The compounds may decrease the expression or activity of the ncRNAs. The compounds or agents may interfere with transcription, by interacting, for example, with the promoter region. The compounds or agents may interfere with mRNA (e.g. , by RNA interference, antisense technologies, etc.). The compounds or agents may interfere with pathways that are upstream or downstream of the biological activity of ncRNAs. In some embodiments, candidate compounds are antisense or interfering RNA agents (e.g. , oligonucleotides) directed against ncRNAs. In other embodiments, candidate compounds are antibodies or small molecules that specifically bind to a ncRNA regulator. Alternatively, or additionally, the candidate compounds are expression products that inhibit thebiological function of the ncRNAs.
In one screening method, candidate compounds are evaluated for their ability to alter ncRNAs expression by contacting a compound with a cell expressing a ncRNA and then assaying for
the effect of the candidate compounds on expression. In some embodiments, the effect of candidate compounds on expression of ncRNAs is assayed for by detecting the level ncRNA expressed by the cell. mRNA expression can be detected by any suitable method.
III. Diagnosis, Prognosis, and Monitoring
The methods, compositions, and kits disclosed herein may be used for the diagnosis, prognosis, and/or monitoring the status or outcome of a cancer in a subject. In some embodiments, the diagnosing, predicting, and/or monitoring the status or outcome of a cancer comprises determining the malignancy or malignant potential of the cancer or tumor. Alternatively, the diagnosing, predicting, and/or monitoring the status or outcome of a cancer comprises determining the stage of the cancer. The diagnosing, predicting, and/or monitoring the status or outcome of a cancer can comprise determining the tumor grade. Alternatively, the diagnosing, predicting, and/or monitoring the status or outcome of a cancer comprises assessing the risk of developing a cancer. In some embodiments, the diagnosing, predicting, and/or monitoring the status or outcome of a cancer includes assessing the risk of cancer recurrence. In some embodiments, diagnosing, predicting, and/or monitoring the status or outcome of a cancer may comprise determining the efficacy of treatment.
In some embodiments, diagnosing, predicting, and/or monitoring the status or outcome of a cancer may comprise determining a therapeutic regimen. Determining a therapeutic regimen may comprise administering an anti-cancer therapeutic. Alternatively, determining the treatment for the cancer may comprise modifying a therapeutic regimen. Modifying a therapeutic regimen may comprise increasing, decreasing, or terminating a therapeutic regimen.
In some instances, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 50%. In other instances, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 60%. The methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 65%. Alternatively, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 70%. In some instances, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 75%. In other instances, the methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 80%. The methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 85%. Alternatively, the methods disclosed herein can diagnose, prognose, and/or monitor the
status or outcome of a cancer in a subject with an accuracy of at least about 90%. The methods disclosed herein can diagnose, prognose, and/or monitor the status or outcome of a cancer in a subject with an accuracy of at least about 95%.
The disclosure also encompasses any of the methods disclosed herein where the sensitivity is at least about 45%. In some embodiments, the sensitivity is at least about 50%. In some embodiments, the sensitivity is at least about 55%. In some embodiments, the sensitivity is at least about 60%. In some embodiments, the sensitivity is at least about 65%. In some embodiments, the sensitivity is at least about 70%. In some embodiments, the sensitivity is at least about 75%. In some embodiments, the sensitivity is at least about 80%. In some embodiments, the sensitivity is at least about 85%. In some embodiments, the sensitivity is at least about 90%. In some embodiments, the sensitivity is at least about 95%.
The disclosure also encompasses any of the methods disclosed herein where the expression level determines the status or outcome of a cancer in the subject with at least about 45% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 50% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 55% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 60% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 65% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 70% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 75% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 80% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 85% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 90% specificity. In some embodiments, the expression level determines the status or outcome of a cancer in the subject with at least about 95% specificity.
Cancer
The systems, compositions and methods disclosed herein may be used to diagnosis, monitor and/or predict the status or outcome of a cancer. Generally, a cancer is characterized by the uncontrolled growth of abnormal cells anywhere in a body. The abnormal cells may be termed cancer cells, malignant cells, or tumor cells. Many cancers and the abnormal cells that compose the cancer tissue are further identified by the name of the tissue that the abnormal cells originated from
(for example, breast cancer, lung cancer, colon cancer, prostate cancer, pancreatic cancer, thyroid cancer). Cancer is not confined to humans; animals and other living organisms can get cancer.
In some instances, the cancer may be malignant. Alternatively, the cancer may be benign. The cancer may be a recurrent and/or refractory cancer. Most cancers can be classified as a carcinoma, sarcoma, leukemia, lymphoma, myeloma, or a central nervous system cancer.
The cancer may be a sarcoma. Sarcomas are cancers of the bone, cartilage, fat, muscle, blood vessels, or other connective or supportive tissue. Sarcomas include, but are not limited to, bone cancer, fibrosarcoma, chondrosarcoma, Ewing's sarcoma, malignant hemangioendothelioma, malignant schwannoma, bilateral vestibular schwannoma, osteosarcoma, soft tissue sarcomas (e.g. alveolar soft part sarcoma, angiosarcoma, cystosarcoma phylloides, dermatofibrosarcoma, desmoid tumor, epithelioid sarcoma, extraskeletal osteosarcoma, fibrosarcoma, hemangiopericytoma, hemangiosarcoma, Kaposi's sarcoma, leiomyosarcoma, liposarcoma, lymphangiosarcoma, lymphosarcoma, malignant fibrous histiocytoma, neurofibrosarcoma, rhabdomyosarcoma, and synovial sarcoma).
Alternatively, the cancer may be a carcinoma. Carcinomas are cancers that begin in the epithelial cells, which are cells that cover the surface of the body, produce hormones, and make up glands. By way of non-limiting example, carcinomas include breast cancer, pancreatic cancer, lung cancer, colon cancer, colorectal cancer, rectal cancer, kidney cancer, bladder cancer, stomach cancer, prostate cancer, liver cancer, ovarian cancer, brain cancer, vaginal cancer, vulvar cancer, uterine cancer, oral cancer, penic cancer, testicular cancer, esophageal cancer, skin cancer, cancer of the fallopian tubes, head and neck cancer, gastrointestinal stromal cancer, adenocarcinoma, cutaneous or intraocular melanoma, cancer of the anal region, cancer of the small intestine, cancer of the endocrine system, cancer of the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal gland, cancer of the urethra, cancer of the renal pelvis, cancer of the ureter, cancer of the
endometrium, cancer of the cervix, cancer of the pituitary gland, neoplasms of the central nervous system (CNS), primary CNS lymphoma, brain stem glioma, and spinal axis tumors. In some instances, the cancer is a skin cancer, such as a basal cell carcinoma, squamous, melanoma, nonmelanoma, or actinic (solar) keratosis. Preferably, the cancer is a prostate cancer. Alternatively, the cancer may be a thyroid cancer. The cancer can be a pancreatic cancer. In some instances, the cancer is a bladder cancer.
In some instances, the cancer is a lung cancer. Lung cancer can start in the airways that branch off the trachea to supply the lungs (bronchi) or the small air sacs of the lung (the alveoli). Lung cancers include non-small cell lung carcinoma (NSCLC), small cell lung carcinoma, and mesotheliomia. Examples of NSCLC include squamous cell carcinoma, adenocarcinoma, and large
cell carcinoma. The mesothelioma may be a cancerous tumor of the lining of the lung and chest cavity (pleura) or lining of the abdomen (peritoneum). The mesothelioma may be due to asbestos exposure. The cancer may be a brain cancer, such as a glioblastoma.
Alternatively, the cancer may be a central nervous system (CNS) tumor. CNS tumors may be classified as gliomas or nongliomas. The glioma may be malignant glioma, high grade glioma, diffuse intrinsic pontine glioma. Examples of gliomas include astrocytomas, oligodendrogliomas (or mixtures of oligodendroglioma and astocytoma elements), and ependymomas. Astrocytomas include, but are not limited to, low-grade astrocytomas, anaplastic astrocytomas, glioblastoma multiforme, pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and subependymal giant cell astrocytoma. Oligodendrogliomas include low-grade oligodendrogliomas (or oligoastrocytomas) and anaplastic oligodendriogliomas. Nongliomas include meningiomas, pituitary adenomas, primary CNS lymphomas, and medulloblastomas. In some instances, the cancer is a meningioma.
The cancer may be leukemia. The leukemia may be an acute lymphocytic leukemia, acute myelocytic leukemia, chronic lymphocytic leukemia, or chronic myelocytic leukemia. Additional types of leukemias include hairy cell leukemia, chronic myelomonocytic leukemia, and juvenile myelomonocytic-leukemia.
In some instances, the cancer is a lymphoma. Lymphomas are cancers of the lymphocytes and may develop from either B or T lymphocytes. The two major types of lymphoma are Hodgkin's lymphoma, previously known as Hodgkin's disease, and non-Hodgkin's lymphoma. Hodgkin's lymphoma is marked by the presence of the Reed-Sternberg cell. Non-Hodgkin's lymphomas are all lymphomas which are not Hodgkin's lymphoma. Non-Hodgkin lymphomas may be indolent lymphomas and aggressive lymphomas. Non-Hodgkin's lymphomas include, but are not limited to, diffuse large B cell lymphoma, follicular lymphoma, mucosa-associated lymphatic tissue lymphoma (MALT), small cell lymphocytic lymphoma, mantle cell lymphoma, Burkitt's lymphoma, mediastinal large B cell lymphoma, Waldenstrom macroglobulinemia, nodal marginal zone B cell lymphoma (NMZL), splenic marginal zone lymphoma (SMZL), extranodal marginal zone B cell lymphoma, intravascular large B cell lymphoma, primary effusion lymphoma, and lymphomatoid granulomatosis.
Cancer Staging
Diagnosing, predicting, or monitoring a status or outcome of a cancer may comprise determining the stage of the cancer. Generally, the stage of a cancer is a description (usually numbers I to IV with IV having more progression) of the extent the cancer has spread. The stage often takes into account the size of a tumor, how deeply it has penetrated, whether it has invaded adjacent organs, how many lymph nodes it has metastasized to (if any), and whether it has spread to distant
organs. Staging of cancer can be used as a predictor of survival, and cancer treatment may be determined by staging. Determining the stage of the cancer may occur before, during, or after treatment. The stage of the cancer may also be determined at the time of diagnosis.
Cancer staging can be divided into a clinical stage and a pathologic stage. Cancer staging may comprise the TNM classification. Generally, the TNM Classification of Malignant Tumours (TNM) is a cancer staging system that describes the extent of cancer in a patient's body. T may describe the size of the tumor and whether it has invaded nearby tissue, N may describe regional lymph nodes that are involved, and M may describe distant metastasis (spread of cancer from one body part to another). In the TNM (Tumor, Node, Metastasis) system, clinical stage and pathologic stage are denoted by a small "c" or "p" before the stage (e.g., CT3N1M0 or pT2N0).
Often, clinical stage and pathologic stage may differ. Clinical stage may be based on all of the available information obtained before a surgery to remove the tumor. Thus, it may include information about the tumor obtained by physical examination, radiologic examination, and endoscopy. Pathologic stage can add additional information gained by examination of the tumor microscopically by a pathologist. Pathologic staging can allow direct examination of the tumor and its spread, contrasted with clinical staging which may be limited by the fact that the information is obtained by making indirect observations at a tumor which is still in the body. The TNM staging system can be used for most forms of cancer.
Alternatively, staging may comprise Ann Arbor staging. Generally, Ann Arbor staging is the staging system for lymphomas, both in Hodgkin's lymphoma (previously called Hodgkin's disease) and Non-Hodgkin lymphoma (abbreviated NHL). The stage may depend on both the place where the malignant tissue is located (as located with biopsy, CT scanning and increasingly positron emission tomography) and on systemic symptoms due to the lymphoma ("B symptoms": night sweats, weight loss of >10% or fevers). The principal stage may be determined by location of the tumor. Stage I may indicate that the cancer is located in a single region, usually one lymph node and the surrounding area. Stage I often may not have outward symptoms. Stage II can indicate that the cancer is located in two separate regions, an affected lymph node or organ and a second affected area, and that both affected areas are confined to one side of the diaphragm - that is, both are above the diaphragm, or both are below the diaphragm. Stage III often indicates that the cancer has spread to both sides of the diaphragm, including one organ or area near the lymph nodes or the spleen. Stage IV may indicate diffuse or disseminated involvement of one or more extralymphatic organs, including any involvement of the liver, bone marrow, or nodular involvement of the lungs.
Modifiers may also be appended to some stages. For example, the letters A, B, E, X, or S can be appended to some stages. Generally, A or B may indicate the absence of constitutional (B-type)
symptoms is denoted by adding an "A" to the stage; the presence is denoted by adding a "B" to the stage. E can be used if the disease is "extranodal" (not in the lymph nodes) or has spread from lymph nodes to adjacent tissue. X is often used if the largest deposit is >10 cm large ("bulky disease"), or whether the mediastinum is wider than 1/3 of the chest on a chest X-ray. S may be used if the disease has spread to the spleen.
The nature of the staging may be expressed with CS or PS. CS may denote that the clinical stage as obtained by doctor's examinations and tests. PS may denote that the pathological stage as obtained by exploratory laparotomy (surgery performed through an abdominal incision) with splenectomy (surgical removal of the spleen).
Therapeutic regimens
Diagnosing, predicting, or monitoring a status or outcome of a cancer may comprise treating a cancer or preventing a cancer progression. In addition, diagnosing, predicting, or monitoring a status or outcome of a cancer may comprise identifying or predicting responders to an anti-cancer therapy. In some instances, diagnosing, predicting, or monitoring may comprise determining a therapeutic regimen. Determining a therapeutic regimen may comprise administering an anti-cancer therapy. Alternatively, determining a therapeutic regimen may comprise modifying, recommending, continuing or discontinuing an anti-cancer regimen. In some instances, if the sample expression patterns are consistent with the expression partem for a known disease or disease outcome, the expression patterns can be used to designate one or more treatment modalities (e.g., therapeutic regimens, anti-cancer regimen). An anti-cancer regimen may comprise one or more anti-cancer therapies. Examples of anti-cancer therapies include targeting cancer therapy (e.g., targeting the non- coding RNAs described herein), surgery, chemotherapy, radiation therapy,
immunotherapy /biological therapy, photodynamic therapy.
In some embodiments, the present disclsoure targets the expression of cancer markers. For example, in some embodiments, the present disclsoure employs compositions comprising oligomeric antisense or RNAi compounds, particularly oligonucleotides (e.g. , those identified in the drug screening methods described above), for use in modulating the function of nucleic acid molecules encoding cancer markers of the present disclsoure, ultimately modulating the amount of cancer marker expressed.
In some embodiments, RNAi is utilized to target non-coding RNAs (e.g., one or more of SEQ
ID NOs: 1-2309). RNAi represents an evolutionary conserved cellular defense for controlling the expression of foreign genes in most eukaryotes, including humans. RNAi is typically triggered by double-stranded RNA (dsRNA) and causes sequence-specific mRNA degradation of single-stranded target RNAs homologous in response to dsRNA. The mediators of mRNA degradation are small
interfering RNA duplexes (siRNAs), which are normally produced from long dsRNA by enzymatic cleavage in the cell. siRNAs are generally approximately twenty-one nucleotides in length (e.g. 21- 23 nucleotides in length), and have a base-paired structure characterized by two nucleotide 3'- overhangs. Following the introduction of a small RNA, or RNAi, into the cell, it is believed the sequence is delivered to an enzyme complex called RISC (RNA-induced silencing complex). RISC recognizes the target and cleaves it with an endonuclease. It is noted that if larger RNA sequences are delivered to a cell, RNase III enzyme (Dicer) converts longer dsRNA into 21-23 nt ds siRNA fragments.
Chemically synthesized siRNAs have become powerful reagents for genome-wide analysis of mammalian gene function in cultured somatic cells. Beyond their value for validation of gene function, siRNAs also hold great potential as gene-specific therapeutic agents (Tuschl and Borkhardt, Molecular Intervent. 2002; 2(3): 158-67, herein incorporated by reference).
The transfection of siRNAs into animal cells results in the potent, long-lasting post- transcriptional silencing of specific genes (Caplen et al, Proc Natl Acad Sci U.S.A. 2001; 98: 9742- 7; Elbashir et al, Nature. 2001 ; 411 :494-8; Elbashir et al, Genes Dev. 2001;15: 188-200; and Elbashir et al, EMBO J. 2001; 20: 6877-88, all of which are herein incorporated by reference). Methods and compositions for performing RNAi with siRNAs are described, for example, in U.S. Pat. 6,506,559, herein incorporated by reference.
siRNAs are extraordinarily effective at lowering the amounts of targeted RNA, and by extension proteins, frequently to undetectable levels. The silencing effect can last several months, and is extraordinarily specific, because one nucleotide mismatch between the target RNA and the central region of the siRNA is frequently sufficient to prevent silencing (Brummelkamp et al, Science 2002; 296:550-3; and Holen et al, Nucleic Acids Res. 2002; 30: 1757-66, both of which are herein incorporated by reference).
An important factor in the design of siRNAs is the presence of accessible sites for siRNA binding. Bahoia et al., (J. Biol. Chem, 2003; 278: 15991-15997; herein incorporated by reference) describe the use of a type of DNA array called a scanning array to find accessible sites in mRNAs for designing effective siRNAs. These arrays comprise oligonucleotides ranging in size from monomers to a certain maximum, usually Comers, synthesized using a physical barrier (mask) by stepwise addition of each base in the sequence. Thus the arrays represent a full oligonucleotide complement of a region of the target gene. Hybridization of the target mRNA to these arrays provides an exhaustive accessibility profile of this region of the target mRNA. Such data are useful in the design of antisense oligonucleotides (ranging from 7mers to 25mers), where it is important to achieve a compromise between oligonucleotide length and binding affinity, to retain efficacy and target specificity (Sohail
et al, Nucleic Acids Res., 2001; 29(10): 2041- 2045). Additional methods and concerns for selecting siRNAs are described for example, in WO 05054270, WO05038054A1, WO03070966A2, J Mol Biol. 2005 May 13;348(4):883-93, J Mol Biol. 2005 May 13;348(4):871-81, and Nucleic Acids Res. 2003 Aug l;31(15):4417-24, each of which is herein incorporated by reference in its entirety. In addition, software (e.g., the MWG online siMAX siRNA design tool) is commercially or publicly available for use in the selection of siRNAs.
In other embodiments, expression of non-coding RNAs (e.g., one or more of SEQ ID NOs: 1- 2309) is modulated using antisense compounds that specifically hybridize with one or more nucleic acids encoding the RNAs. The specific hybridization of an oligomeric compound with its target nucleic acid interferes with the normal function of the nucleic acid. This modulation of function of a target nucleic acid by compounds that specifically hybridize to it is generally referred to as
"antisense." The functions of DNA to be interfered with include replication and transcription. The functions of RNA to be interfered with include all vital functions such as, for example, translocation of the RNA to the site of protein translation, translation of protein from the RNA, splicing of the RNA to yield one or more mRNA species, and catalytic activity that may be engaged in or facilitated by the RNA. The overall effect of such interference with target nucleic acid function is modulation of the expression of cancer markers of the present disclsoure. In the context of the present disclsoure, "modulation" means either an increase (stimulation) or a decrease (inhibition) in the expression of a gene. For example, expression may be inhibited to potentially prevent tumor proliferation.
It is preferred to target specific nucleic acids for antisense. "Targeting" an antisense compound to a particular nucleic acid, in the context of the present disclsoure, is a multistep process. The process usually begins with the identification of a nucleic acid sequence whose function is to be modulated. This may be, for example, a cellular gene (or mRNA transcribed from the gene) whose expression is associated with a particular disorder or disease state, or a nucleic acid molecule from an infectious agent. In the present disclsoure, the target is a nucleic acid molecule encoding a cancer marker of the present disclsoure. The targeting process also includes determination of a site or sites within this gene for the antisense interaction to occur such that the desired effect, e.g., detection or modulation of expression of the protein, will result. Within the context of the present disclsoure, a preferred intragenic site is the region encompassing the translation initiation or termination codon of the open reading frame (ORF) of the gene. Since the translation initiation codon is typically 5'-AUG (in transcribed mRNA molecules; 5'-ATG in the corresponding DNA molecule), the translation initiation codon is also referred to as the "AUG codon," the "start codon" or the "AUG start codon". A minority of genes have a translation initiation codon having the RNA sequence 5'-GUG, 5'-UUG
or 5'-CUG, and 5'-AUA, 5'-ACG and 5'-CUG have been shown to function in vivo. Thus, the terms "translation initiation codon" and "start codon" can encompass many codon sequences, even though the initiator amino acid in each instance is typically methionine (in eukaryotes) or formylmethionine (in prokaryotes). Eukaryotic and prokaryotic genes may have two or more alternative start codons, any one of which may be preferentially utilized for translation initiation in a particular cell type or tissue, or under a particular set of conditions. In the context of the present disclsoure, "start codon" and "translation initiation codon" refer to the codon or codons that are used in vivo to initiate translation of an RNA (e.g., one or more of SEQ ID NOs: 1-2309).
Translation termination codon (or "stop codon") of a gene may have one of three sequences (/. e. , 5'-UAA, 5'-UAG and 5'-UGA; the corresponding DNA sequences are 5'-TAA, 5'-TAG and
5'-TGA, respectively). The terms "start codon region" and "translation initiation codon region" refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction (i.e. , 5' or 3') from a translation initiation codon. Similarly, the terms "stop codon region" and "translation termination codon region" refer to a portion of such an mRNA or gene that encompasses from about 25 to about 50 contiguous nucleotides in either direction (i.e. , 5' or 3') from a translation termination codon.
The open reading frame (ORF) or "coding region," which refers to the region between the translation initiation codon and the translation termination codon, is also a region that may be targeted effectively. Other target regions include the 5' untranslated region (5' UTR), referring to the portion of an mRNA in the 5' direction from the translation initiation codon, and thus including nucleotides between the 5' cap site and the translation initiation codon of an mRNA or corresponding nucleotides on the gene, and the 3' untranslated region (3' UTR), referring to the portion of an mRNA in the 3' direction from the translation termination codon, and thus including nucleotides between the translation termination codon and 3' end of an mRNA or corresponding nucleotides on the gene. The 5' cap of an mRNA comprises an N7-methylated guanosine residue joined to the 5'-most residue of the mRNA via a 5'-5' triphosphate linkage. The 5' cap region of an mRNA is considered to include the 5' cap structure itself as well as the first 50 nucleotides adjacent to the cap. The cap region may also be a preferred target region.
Although some eukaryotic mRNA transcripts are directly translated, many contain one or more regions, known as "introns," that are excised from a transcript before it is translated. The remaining (and therefore translated) regions are known as "exons" and are spliced together to form a continuous mRNA sequence. mRNA splice sites (i.e. , intron-exon junctions) may also be preferred target regions, and are particularly useful in situations where aberrant splicing is implicated in disease, or where an overproduction of a particular mRNA splice product is implicated in disease. It
has also been found that introns can also be effective, and therefore preferred, target regions for antisense compounds targeted, for example, to DNA or pre-mRNA.
In some embodiments, target sites for antisense inhibition are identified using commercially available software programs (e.g., Biognostik, Gottingen, Germany; SysArris Software, Bangalore, India; Antisense Research Group, University of Liverpool, Liverpool, England; GeneTrove,
Carlsbad, CA). In other embodiments, target sites for antisense inhibition are identified using the accessible site method described in PCT Publ. No. WO0198537A2, herein incorporated by reference.
Once one or more target sites have been identified, oligonucleotides are chosen that are sufficiently complementary to the target (i.e. , hybridize sufficiently well and with sufficient specificity) to give the desired effect. For example, in preferred embodiments of the present disclsoure, antisense oligonucleotides are targeted to or near the start codon.
In the context of this disclsoure, "hybridization," with respect to antisense compositions and methods, means hydrogen bonding, which may be Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen bonding, between complementary nucleoside or nucleotide bases. For example, adenine and thymine are complementary nucleobases that pair through the formation of hydrogen bonds. It is understood that the sequence of an antisense compound need not be 100% complementary to that of its target nucleic acid to be specifically hybridizable. An antisense compound is specifically hybridizable when binding of the compound to the target DNA or RNA molecule interferes with the normal function of the target DNA or RNA to cause a loss of utility, and there is a sufficient degree of complementarity to avoid non-specific binding of the antisense compound to non-target sequences under conditions in which specific binding is desired (i.e., under physiological conditions in the case of in vivo assays or therapeutic treatment, and in the case of in vitro assays, under conditions in which the assays are performed).
The specificity and sensitivity of antisense is also applied for therapeutic uses. For example, antisense oligonucleotides have been employed as therapeutic moieties in the treatment of disease states in animals and man. Antisense oligonucleotides have been safely and effectively administered to humans and numerous clinical trials are presently underway. It is thus established that oligonucleotides are useful therapeutic modalities that can be configured to be useful in treatment regimes for treatment of cells, tissues, and animals, especially humans.
While antisense oligonucleotides are a preferred form of antisense compound, the present disclsoure comprehends other oligomeric antisense compounds, including but not limited to oligonucleotide mimetics such as are described below. The antisense compounds in accordance with this disclsoure preferably comprise from about 8 to about 30 nucleobases (i.e., from about 8 to about 30 linked bases), although both longer and shorter sequences may find use with the present
disclsoure. Particularly preferred antisense compounds are antisense oligonucleotides, even more preferably those comprising from about 12 to about 25 nucleobases.
Specific examples of preferred antisense compounds useful with the present disclsoure include oligonucleotides containing modified backbones or non-natural intemucleoside linkages. As defined in this specification, oligonucleotides having modified backbones include those that retain a phosphorus atom in the backbone and those that do not have a phosphorus atom in the backbone. For the purposes of this specification, modified oligonucleotides that do not have a phosphorus atom in their intemucleoside backbone can also be considered to be oligonucleosides.
Preferred modified oligonucleotide backbones include, for example, phosphorothioates, chiral phosphorothioates, phosphorodithioates, phosphotriesters, aminoalkylphosphotriesters, methyl and other alkyl phosphonates including 3'-alkylene phosphonates and chiral phosphonates, phosphinates, phosphoramidates including 3 '-amino phosphoramidate and
aminoalkylphosphoramidates, thionophosphoramidates, thionoalkylphosphonates,
thionoalkylphosphotriesters, and boranophosphates having normal 3'-5' linkages, 2'-5' linked analogs of these, and those having inverted polarity wherein the adjacent pairs of nucleoside units are linked 3'-5' to 5'-3' or 2'-5' to 5'-2'. Various salts, mixed salts and free acid forms are also included.
The present disclsoure contemplates the use of any genetic manipulation for use in modulating the expression of non-coding RNAs (e.g., one or more of SEQ ID NOs: 1-2309).
Examples of genetic manipulation include, but are not limited to, gene knockout (e.g., removing the gene encoding the RNA from the chromosome using, for example, recombination), expression of antisense constructs with or without inducible promoters, and the like. Delivery of nucleic acid construct to cells in vitro or in vivo may be conducted using any suitable method. A suitable method is one that introduces the nucleic acid construct into the cell such that the desired event occurs (e.g., expression of an antisense construct). Genetic therapy may also be used to deliver siRNA or other interfering molecules that are expressed in vivo (e.g., upon stimulation by an inducible promoter (e.g., an androgen-responsive promoter)).
Introduction of molecules carrying genetic information into cells is achieved by any of various methods including, but not limited to, directed injection of naked DNA constructs, bombardment with gold particles loaded with said constructs, and macromolecule mediated gene transfer using, for example, liposomes, biopolymers, and the like. Preferred methods use gene delivery vehicles derived from viruses, including, but not limited to, adenoviruses, retroviruses, vaccinia viruses, and adeno-associated viruses. Because of the higher efficiency as compared to retroviruses, vectors derived from adenoviruses are the preferred gene delivery vehicles for transferring nucleic acid molecules into host cells in vivo. Adenoviral vectors have been shown to
provide very efficient in vivo gene transfer into a variety of solid tumors in animal models and into human solid tumor xenografts in immune-deficient mice. Examples of adenoviral vectors and methods for gene transfer are described in PCT publications WO 00/12738 and WO 00/09675 and U.S. Pat. Appl. Nos. 6,033,908, 6,019,978, 6,001,557, 5,994,132, 5,994,128, 5,994,106, 5,981,225, 5,885,808, 5,872,154, 5,830,730, and 5,824,544, each of which is herein incorporated by reference in its entirety.
Vectors may be administered to subject in a variety of ways. For example, in some embodiments of the present disclsoure, vectors are administered into tumors or tissue associated with tumors using direct injection. In other embodiments, administration is via the blood or lymphatic circulation (See e.g., PCT publication 99/02685 herein incorporated by reference in its entirety). Exemplary dose levels of adenoviral vector are preferably 108 to 1011 vector particles added to the perfusate.
Surgical oncology uses surgical methods to diagnose, stage, and treat cancer, and to relieve certain cancer-related symptoms. Surgery may be used to remove the tumor (e.g., excisions, resections, debulking surgery), reconstruct a part of the body (e.g., restorative surgery), and/or to relieve symptoms such as pain (e.g., palliative surgery). Surgery may also include cryosurgery.
Cryosurgery (also called cryotherapy) may use extreme cold produced by liquid nitrogen (or argon gas) to destroy abnormal tissue. Cryosurgery can be used to treat external tumors, such as those on the skin. For external tumors, liquid nitrogen can be applied directly to the cancer cells with a cotton swab or spraying device. Cryosurgery may also be used to treat tumors inside the body (internal tumors and tumors in the bone). For internal tumors, liquid nitrogen or argon gas may be circulated through a hollow instrument called a cryoprobe, which is placed in contact with the tumor. An ultrasound or MRI may be used to guide the cryoprobe and monitor the freezing of the cells, thus limiting damage to nearby healthy tissue. A ball of ice crystals may form around the probe, freezing nearby cells. Sometimes more than one probe is used to deliver the liquid nitrogen to various parts of the tumor. The probes may be put into the tumor during surgery or through the skin (percutaneously). After cryosurgery, the frozen tissue thaws and may be naturally absorbed by the body (for internal tumors), or may dissolve and form a scab (for external tumors).
Chemotherapeutic agents may also be used for the treatment of cancer. Examples of chemotherapeutic agents include alkylating agents, anti-metabolites, plant alkaloids and terpenoids, vinca alkaloids, podophyllotoxin, taxanes, topoisomerase inhibitors, and cytotoxic antibiotics.
Cisplatin, carboplatin, and oxaliplatin are examples of alkylating agents. Other alkylating agents include mechlorethamine, cyclophosphamide, chlorambucil, ifosfamide. Alkylating agens may impair cell function by forming covalent bonds with the amino, carboxyl, sulfhydryl, and phosphate
groups in biologically important molecules. Alternatively, alkylating agents may chemically modify a cell's DNA.
Anti-metabolites are another example of chemotherapeutic agents. Anti-metabolites may masquerade as purines or pyrimidines and may prevent purines and pyrimidines from becoming incorporated in to DNA during the "S" phase (of the cell cycle), thereby stopping normal development and division. Antimetabolites may also affect RNA synthesis. Examples of metabolites include azathioprine and mercaptopurine.
Alkaloids may be derived from plants and block cell division may also be used for the treatment of cancer. Alkyloids may prevent microtubule function. Examples of alkaloids are vinca alkaloids and taxanes. Vinca alkaloids may bind to specific sites on tubulin and inhibit the assembly of tubulin into microtubules (M phase of the cell cycle). The vinca alkaloids may be derived from the Madagascar periwinkle, Catharanthus roseus (formerly known as Vinca rosea). Examples of vinca alkaloids include, but are not limited to, vincristine, vinblastine, vinorelbine, or vindesine. Taxanes are diterpenes produced by the plants of the genus Taxus (yews). Taxanes may be derived from natural sources or synthesized artificially. Taxanes include paclitaxel (Taxol) and docetaxel
(Taxotere). Taxanes may disrupt microtubule function. Microtubules are essential to cell division, and taxanes may stabilize GDP-bound tubulin in the microtubule, thereby inhibiting the process of cell division. Thus, in essence, taxanes may be mitotic inhibitors. Taxanes may also be
radiosensitizing and often contain numerous chiral centers.
Alternative chemotherapeutic agents include podophyllotoxin. Podophyllotoxin is a plant- derived compound that may help with digestion and may be used to produce cytostatic drugs such as etoposide and teniposide. They may prevent the cell from entering the Gl phase (the start of DNA replication) and the replication of DNA (the S phase).
Topoisomerases are essential enzymes that maintain the topology of DNA. Inhibition of type I or type II topoisomerases may interfere with both transcription and replication of DNA by upsetting proper DNA supercoiling. Some chemotherapeutic agents may inhibit topoisomerases. For example, some type I topoisomerase inhibitors include camptothecins: irinotecan and topotecan. Examples of type II inhibitors include amsacrine, etoposide, etoposide phosphate, and teniposide.
Another example of chemotherapeutic agents is cytotoxic antibiotics. Cytotoxic antibiotics are a group of antibiotics that are used for the treatment of cancer because they may interfere with DNA replication and/or protein synthesis. Cytotoxic antiobiotics include, but are not limited to, actinomycin, anthracyclines, doxorubicin, daunorubicin, valrubicin, idarubicin, epirubicin, bleomycin, plicamycin, and mitomycin.
In some instances, the anti-cancer treatment may comprise radiation therapy. Radiation can come from a machine outside the body (external-beam radiation therapy) or from radioactive material placed in the body near cancer cells (internal radiation therapy, more commonly called brachytherapy). Systemic radiation therapy uses a radioactive substance, given by mouth or into a vein that travels in the blood to tissues throughout the body.
External-beam radiation therapy may be delivered in the form of photon beams (either x-rays or gamma rays). A photon is the basic unit of light and other forms of electromagnetic radiation. An example of external-beam radiation therapy is called 3-dimensional conformal radiation therapy (3D- CRT). 3D-CRT may use computer software and advanced treatment machines to deliver radiation to very precisely shaped target areas. Many other methods of external -beam radiation therapy are currently being tested and used in cancer treatment. These methods include, but are not limited to, intensity-modulated radiation therapy (IMRT), image-guided radiation therapy (IGRT), Stereotactic radiosurgery (SRS), Stereotactic body radiation therapy (SBRT), and proton therapy.
Intensity-modulated radiation therapy (IMRT) is an example of external-beam radiation and may use hundreds of tiny radiation beam-shaping devices, called collimators, to deliver a single dose of radiation. The collimators can be stationary or can move during treatment, allowing the intensity of the radiation beams to change during treatment sessions. This kind of dose modulation allows different areas of a tumor or nearby tissues to receive different doses of radiation. IMRT is planned in reverse (called inverse treatment planning). In inverse treatment planning, the radiation doses to different areas of the tumor and surrounding tissue are planned in advance, and then a high-powered computer program calculates the required number of beams and angles of the radiation treatment. In contrast, during traditional (forward) treatment planning, the number and angles of the radiation beams are chosen in advance and computers calculate how much dose may be delivered from each of the planned beams. The goal of IMRT is to increase the radiation dose to the areas that need it and reduce radiation exposure to specific sensitive areas of surrounding normal tissue.
Another example of external-beam radiation is image-guided radiation therapy (IGRT). In IGRT, repeated imaging scans (CT, MRI, or PET) may be performed during treatment. These imaging scans may be processed by computers to identify changes in a tumor's size and location due to treatment and to allow the position of the patient or the planned radiation dose to be adjusted during treatment as needed. Repeated imaging can increase the accuracy of radiation treatment and may allow reductions in the planned volume of tissue to be treated, thereby decreasing the total radiation dose to normal tissue.
Tomotherapy is a type of image-guided IMRT. A tomotherapy machine is a hybrid between a CT imaging scanner and an external-beam radiation therapy machine. The part of the tomotherapy
machine that delivers radiation for both imaging and treatment can rotate completely around the patient in the same manner as a normal CT scanner. Tomotherapy machines can capture CT images of the patient's tumor immediately before treatment sessions, to allow for very precise tumor targeting and sparing of normal tissue.
Stereotactic radiosurgery (SRS) can deliver one or more high doses of radiation to a small tumor. SRS uses extremely accurate image-guided tumor targeting and patient positioning.
Therefore, a high dose of radiation can be given without excess damage to normal tissue. SRS can be used to treat small tumors with well-defined edges. It is most commonly used in the treatment of brain or spinal tumors and brain metastases from other cancer types. For the treatment of some brain metastases, patients may receive radiation therapy to the entire brain (called whole-brain radiation therapy) in addition to SRS. SRS requires the use of a head frame or other device to immobilize the patient during treatment to ensure that the high dose of radiation is delivered accurately.
Stereotactic body radiation therapy (SBRT) delivers radiation therapy in fewer sessions, using smaller radiation fields and higher doses than 3D-CRT in most cases. SBRT may treat tumors that lie outside the brain and spinal cord. Because these tumors are more likely to move with the normal motion of the body, and therefore cannot be targeted as accurately as tumors within the brain or spine, SBRT is usually given in more than one dose. SBRT can be used to treat small, isolated tumors, including cancers in the lung and liver. SBRT systems may be known by their brand names, such as the CyberKnife®.
In proton therapy, external-beam radiation therapy may be delivered by proton. Protons are a type of charged particle. Proton beams differ from photon beams mainly in the way they deposit energy in living tissue. Whereas photons deposit energy in small packets all along their path through tissue, protons deposit much of their energy at the end of their path (called the Bragg peak) and deposit less energy along the way. Use of protons may reduce the exposure of normal tissue to radiation, possibly allowing the delivery of higher doses of radiation to a tumor.
Other charged particle beams such as electron beams may be used to irradiate superficial tumors, such as skin cancer or tumors near the surface of the body, but they cannot travel very far through tissue.
Internal radiation therapy (brachytherapy) is radiation delivered from radiation sources (radioactive materials) placed inside or on the body. Several brachytherapy techniques are used in cancer treatment. Interstitial brachytherapy may use a radiation source placed within tumor tissue, such as within a prostate tumor. Intracavitary brachytherapy may use a source placed within a surgical cavity or a body cavity, such as the chest cavity, near a tumor. Episcleral brachytherapy, which may be used to treat melanoma inside the eye, may use a source that is attached to the eye. In
brachytherapy, radioactive isotopes can be sealed in tiny pellets or "seeds." These seeds may be placed in patients using delivery devices, such as needles, catheters, or some other type of carrier. As the isotopes decay naturally, they give off radiation that may damage nearby cancer cells.
Brachytherapy may be able to deliver higher doses of radiation to some cancers than external-beam radiation therapy while causing less damage to normal tissue.
Brachytherapy can be given as a low-dose-rate or a high-dose-rate treatment. In low-dose- rate treatment, cancer cells receive continuous low-dose radiation from the source over a period of several days. In high-dose-rate treatment, a robotic machine attached to delivery tubes placed inside the body may guide one or more radioactive sources into or near a tumor, and then removes the sources at the end of each treatment session. High-dose-rate treatment can be given in one or more treatment sessions. An example of a high-dose-rate treatment is the MammoSite® system.
Bracytherapy may be used to treat patients with breast cancer who have undergone breast-conserving surgery.
The placement of brachytherapy sources can be temporary or permanent. For permament brachytherapy, the sources may be surgically sealed within the body and left there, even after all of the radiation has been given off. In some instances, the remaining material (in which the radioactive isotopes were sealed) does not cause any discomfort or harm to the patient. Permanent brachytherapy is a type of low-dose-rate brachytherapy. For temporary brachytherapy, tubes (catheters) or other carriers are used to deliver the radiation sources, and both the carriers and the radiation sources are removed after treatment. Temporary brachytherapy can be either low-dose-rate or high-dose-rate treatment. Brachytherapy may be used alone or in addition to external-beam radiation therapy to provide a "boost" of radiation to a tumor while sparing surrounding normal tissue.
In systemic radiation therapy, a patient may swallow or receive an injection of a radioactive substance, such as radioactive iodine or a radioactive substance bound to a monoclonal antibody. Radioactive iodine (1 1I) is a type of systemic radiation therapy commonly used to help treat cancer, such as thyroid cancer. Thyroid cells naturally take up radioactive iodine. For systemic radiation therapy for some other types of cancer, a monoclonal antibody may help target the radioactive substance to the right place. The antibody joined to the radioactive substance travels through the blood, locating and killing tumor cells. For example, the drug ibritumomab tiuxetan (Zevalin®) may be used for the treatment of certain types of B-cell non-Hodgkin lymphoma (NHL). The antibody part of this drug recognizes and binds to a protein found on the surface of B lymphocytes. The combination drug regimen of tositumomab and iodine 1 T tositumomab (Bexxar®) may be used for the treatment of certain types of cancer, such as NHL. In this regimen, nonradioactive tositumomab antibodies may be given to patients first, followed by treatment with tositumomab antibodies that
have 1 T attached. Tositumomab may recognize and bind to the same protein on B lymphocytes as ibritumomab. The nonradioactive form of the antibody may help protect normal B lymphocytes from being damaged by radiation from 1311.
Some systemic radiation therapy drugs relieve pain from cancer that has spread to the bone (bone metastases). This is a type of palliative radiation therapy. The radioactive drugs samarium- 153-lexidronam (Quadramet®) and strontium-89 chloride (Metastron®) are examples of
radiopharmaceuticals may be used to treat pain from bone metastases.
Biological therapy (sometimes called immunotherapy, biotherapy, or biological response modifier (BRM) therapy) uses the body's immune system, either directly or indirectly, to fight cancer or to lessen the side effects that may be caused by some cancer treatments. Biological therapies include interferons, interleukins, colony-stimulating factors, monoclonal antibodies, vaccines, gene therapy, and nonspecific immunomodulating agents.
Interferons (IFNs) are types of cytokines that occur naturally in the body. Interferon alpha, interferon beta, and interferon gamma are examples of interferons that may be used in cancer treatment.
Like interferons, interleukins (ILs) are cytokines that occur naturally in the body and can be made in the laboratory. Many interleukins have been identified for the treatment of cancer. For example, interleukin-2 (IL-2 or aldesleukin), interleukin 7, and interleukin 12 have may be used as an anti-cancer treatment. IL-2 may stimulate the growth and activity of many immune cells, such as lymphocytes, that can destroy cancer cells. Interleukins may be used to treat a number of cancers, including leukemia, lymphoma, and brain, colorectal, ovarian, breast, kidney and prostate cancers.
Colony-stimulating factors (CSFs) (sometimes called hematopoietic growth factors) may also be used for the treatment of cancer. Some examples of CSFs include, but are not limited to, G-CSF (filgrastim) and GM-CSF (sargramostim). CSFs may promote the division of bone marrow stem cells and their development into white blood cells, platelets, and red blood cells. Bone marrow is critical to the body's immune system because it is the source of all blood cells. Because anticancer drugs can damage the body's ability to make white blood cells, red blood cells, and platelets, stimulation of the immune system by CSFs may benefit patients undergoing other anti-cancer treatment, thus CSFs may be combined with other anti-cancer therapies, such as chemotherapy. CSFs may be used to treat a large variety of cancers, including lymphoma, leukemia, multiple myeloma, melanoma, and cancers of the brain, lung, esophagus, breast, uterus, ovary, prostate, kidney, colon, and rectum.
Another type of biological therapy includes monoclonal antibodies (MOABs or MoABs). These antibodies may be produced by a single type of cell and may be specific for a particular antigen. To create MOABs, human cancer cells may be injected into mice. In response, the mouse
immune system can make antibodies against these cancer cells. The mouse plasma cells that produce antibodies may be isolated and fused with laboratory-grown cells to create "hybrid" cells called hybridomas. Hybridomas can indefinitely produce large quantities of these pure antibodies, or MOABs. MOABs may be used in cancer treatment in a number of ways. For instance, MOABs that react with specific types of cancer may enhance a patient's immune response to the cancer. MOABs can be programmed to act against cell growth factors, thus interfering with the growth of cancer cells.
MOABs may be linked to other anti-cancer therapies such as chemotherapeutics,
radioisotopes (radioactive substances), other biological therapies, or other toxins. When the antibodies latch onto cancer cells, they deliver these anti-cancer therapies directly to the tumor, helping to destroy it. MOABs carrying radioisotopes may also prove useful in diagnosing certain cancers, such as colorectal, ovarian, and prostate.
Rituxan® (rituximab) and Herceptin® (trastuzumab) are examples of MOABs that may be used as a biological therapy. Rituxan may be used for the treatment of non-Hodgkin lymphoma. Herceptin can be used to treat metastatic breast cancer in patients with tumors that produce excess amounts of a protein called HER2. Alternatively, MOABs may be used to treat lymphoma, leukemia, melanoma, and cancers of the brain, breast, lung, kidney, colon, rectum, ovary, prostate, and other areas.
Cancer vaccines are another form of biological therapy. Cancer vaccines may be designed to encourage the patient's immune system to recognize cancer cells. Cancer vaccines may be designed to treat existing cancers (therapeutic vaccines) or to prevent the development of cancer (prophylactic vaccines). Therapeutic vaccines may be injected in a person after cancer is diagnosed. These vaccines may stop the growth of existing tumors, prevent cancer from recurring, or eliminate cancer cells not killed by prior treatments. Cancer vaccines given when the tumor is small may be able to eradicate the cancer. On the other hand, prophylactic vaccines are given to healthy individuals before cancer develops. These vaccines are designed to stimulate the immune system to attack viruses that can cause cancer. By targeting these cancer-causing viruses, development of certain cancers may be prevented. For example, cervarix and gardasil are vaccines to treat human papilloma virus and may prevent cervical cancer. Therapeutic vaccines may be used to treat melanoma, lymphoma, leukemia, and cancers of the brain, breast, lung, kidney, ovary, prostate, pancreas, colon, and rectum. Cancer vaccines can be used in combination with other anti-cancer therapies.
Gene therapy is another example of a biological therapy. Gene therapy may involve introducing genetic material into a person's cells to fight disease. Gene therapy methods may improve a patient's immune response to cancer. For example, a gene may be inserted into an immune
cell to enhance its ability to recognize and attack cancer cells. In another approach, cancer cells may be injected with genes that cause the cancer cells to produce cytokines and stimulate the immune system.
In some instances, biological therapy includes nonspecific immunomodulating agents.
Nonspecific immunomodulating agents are substances that stimulate or indirectly augment the immune system. Often, these agents target key immune system cells and may cause secondary responses such as increased production of cytokines and immunoglobulins. Two nonspecific immunomodulating agents used in cancer treatment are bacillus Calmette-Guerin (BCG) and levamisole. BCG may be used in the treatment of superficial bladder cancer following surgery. BCG may work by stimulating an inflammatory, and possibly an immune, response. A solution of BCG may be instilled in the bladder. Levamisole is sometimes used along with fluorouracil (5-FU) chemotherapy in the treatment of stage III (Dukes' C) colon cancer following surgery. Levamisole may act to restore depressed immune function.
Photodynamic therapy (PDT) is an anti-cancer treatment that may use a drug, called a photosensitizer or photosensitizing agent, and a particular type of light. When photos ensitizers are exposed to a specific wavelength of light, they may produce a form of oxygen that kills nearby cells. A photosensitizer may be activated by light of a specific wavelength. This wavelength determines how far the light can travel into the body. Thus, photos ensitizers and wavelengths of light may be used to treat different areas of the body with PDT.
In the first step of PDT for cancer treatment, a photosensitizing agent may be injected into the bloodstream. The agent may be absorbed by cells all over the body but may stay in cancer cells longer than it does in normal cells. Approximately 24 to 72 hours after injection, when most of the agent has left normal cells but remains in cancer cells, the tumor can be exposed to light. The photosensitizer in the tumor can absorb the light and produces an active form of oxygen that destroys nearby cancer cells. In addition to directly killing cancer cells, PDT may shrink or destroy tumors in two other ways. The photosensitizer can damage blood vessels in the tumor, thereby preventing the cancer from receiving necessary nutrients. PDT may also activate the immune system to attack the tumor cells.
The light used for PDT can come from a laser or other sources. Laser light can be directed through fiber optic cables (thin fibers that transmit light) to deliver light to areas inside the body. For example, a fiber optic cable can be inserted through an endoscope (a thin, lighted tube used to look at tissues inside the body) into the lungs or esophagus to treat cancer in these organs. Other light sources include light-emitting diodes (LEDs), which may be used for surface tumors, such as skin
cancer. PDT is usually performed as an outpatient procedure. PDT may also be repeated and may be used with other therapies, such as surgery, radiation, or chemotherapy.
Extracorporeal photopheresis (ECP) is a type of PDT in which a machine may be used to collect the patient's blood cells. The patient's blood cells may be treated outside the body with a photosensitizing agent, exposed to light, and then returned to the patient. ECP may be used to help lessen the severity of skin symptoms of cutaneous T-cell lymphoma that has not responded to other therapies. ECP may be used to treat other blood cancers, and may also help reduce rejection after transplants.
Additionally, photosensitizing agent, such as porfimer sodium or Photofrin®, may be used in PDT to treat or relieve the symptoms of esophageal cancer and non-small cell lung cancer. Porfimer sodium may relieve symptoms of esophageal cancer when the cancer obstructs the esophagus or when the cancer cannot be satisfactorily treated with laser therapy alone. Porfimer sodium may be used to treat non-small cell lung cancer in patients for whom the usual treatments are not appropriate, and to relieve symptoms in patients with non-small cell lung cancer that obstructs the airways.
Porfimer sodium may also be used for the treatment of precancerous lesions in patients with Barrett esophagus, a condition that can lead to esophageal cancer.
Laser therapy may use high-intensity light to treat cancer and other illnesses. Lasers can be used to shrink or destroy tumors or precancerous growths. Lasers are most commonly used to treat superficial cancers (cancers on the surface of the body or the lining of internal organs) such as basal cell skin cancer and the very early stages of some cancers, such as cervical, penile, vaginal, vulvar, and non-small cell lung cancer.
Lasers may also be used to relieve certain symptoms of cancer, such as bleeding or obstruction. For example, lasers can be used to shrink or destroy a tumor that is blocking a patient's trachea (windpipe) or esophagus. Lasers also can be used to remove colon polyps or tumors that are blocking the colon or stomach.
Laser therapy is often given through a flexible endoscope (a thin, lighted tube used to look at tissues inside the body). The endoscope is fitted with optical fibers (thin fibers that transmit light). It is inserted through an opening in the body, such as the mouth, nose, anus, or vagina. Laser light is then precisely aimed to cut or destroy a tumor.
Laser-induced interstitial thermotherapy (LITT), or interstitial laser photocoagulation, also uses lasers to treat some cancers. LITT is similar to a cancer treatment called hyperthermia, which uses heat to shrink tumors by damaging or killing cancer cells. During LITT, an optical fiber is inserted into a tumor. Laser light at the tip of the fiber raises the temperature of the tumor cells and damages or destroys them. LITT is sometimes used to shrink tumors in the liver.
Laser therapy can be used alone, but most often it is combined with other treatments, such as surgery, chemotherapy, or radiation therapy. In addition, lasers can seal nerve endings to reduce pain after surgery and seal lymph vessels to reduce swelling and limit the spread of tumor cells.
Lasers used to treat cancer may include carbon dioxide (C02) lasers, argon lasers, and neodymium:yttrium-aluminum-gamet (Nd:YAG) lasers. Each of these can shrink or destroy tumors and can be used with endoscopes. C02 and argon lasers can cut the skin's surface without going into deeper layers. Thus, they can be used to remove superficial cancers, such as skin cancer. In contrast, the Nd:YAG laser is more commonly applied through an endoscope to treat internal organs, such as the uterus, esophagus, and colon. Nd:YAG laser light can also travel through optical fibers into specific areas of the body during LITT. Argon lasers are often used to activate the drugs used in PDT.
EXPERIMENTAL
The following examples are provided in order to demonstrate and further illustrate certain preferred embodiments and aspects of the present disclosure and are not to be construed as limiting the scope thereof.
Example 1
Methods
High performance computing
Computational analysis was performed using the Flux high-performance computer cluster hosted by the Advanced Research Computing (ARC) at the University of Michigan.
RNA-Seq Data Processing
A comprehensive RNA-Seq analysis pipeline was employed on all samples (Fig. 5b). The analysis pipeline provided sequence quality metrics, filtering of contaminant reads, fragment size estimation, strand-specific library type estimation, spliced alignment of reads to the human reference genome (version hgl9/GrCh37), alignment performance metrics, generation of visualization tracks for genome browsers, and ab initio transcript assembly. The third-party tools used to process RNA- Seq data were selected based on computational performance, ease-of-use, user and community support, and experience.
Software versions were managed effectively using the Modules Environment Management system. Computational analysis was performed in a 64-bit Linux environment (Red Hat Enterprise Linux 6). Pre-compiled 64- bit Linux binaries were downloaded when available.
Initial sequence quality control metrics were calculated using FASTQC. Next, filtering was performed to remove reads mapping to mitochondrial DNA, ribosomal RNA, poly -A, poly-C,
Illumina sequencing adaptors, and the spiked-in phiX174 viral genome. Sequences were downloaded from the Illumina iGenomes server (2012, March 9). Mapping was performed using bowtie2 (2.0.2).
The fragment size distribution (for paired-end libraries) and fragment layout of each library was determined automatically by mapping a subset of the reads to a reference consisting of the 15,868 unique Ensembl v69 exons larger than 500bp that had no other overlapping features on either strand. These exons represent contiguous genomic regions where both paired-end reads from a single fragment could confidently be aligned. An alignment index was prepared from this reference using the bowtie-build utility.
Reads were mapped using Tophat2 (2.0.6 and 2.0.8) using default parametersl. Reference genome annotation files were downloaded from the Illumina iGenomes FTP server. A human genome reference was constructed from UCSC version hgl9 chromosomes 1-22, X, Y, and mitochondrial DNA. References from alternate haplotype alleles were omitted. Alignment index files for Bowtie versions 0.12.8 and 2.0.2 were built from this reference using the bowtie-build and bowtie2-build programs, respectively. The Ensembl version 69 transcriptome reference gene set was downloaded from the Ensembl FTP server. Chromosome names were converted from GRCh37 format to UCSC format (e.g. "1" converted to "chrl"). Genes found on alternate haplotype alleles were omitted. The cuffcompare utility was used as specified in the Cufflinks user's manual to assign promoter and transcription start site attributes to the gene features in the Ensembl reference.
Alignment index files for Bowtie versions 0.12.8 and 2.0.2 were prepared from this reference using the -transcriptome index option in Tophat version 2.0.6.
Sequence alignment metrics were computed using the Picard tools CollectMultipleMetrics and CollectRnaSeqMetrics. The Picard CollectRnaSeqMetrics diagnostic utility required gene annotation and ribosomal interval files as input. The "refFlat" table provided by the Illumina iGenomes download package (2012, March 9) was used as the gene annotation reference. Ribosomal DNA intervals were curated from the RepeatMasker table downloaded from the UCSC table browser (Karolchik, D. et al. Nucleic acids research 32, D493-496, (2004)). This table of repeat elements was originally provided for hgl9 by UCSC on 4/27/2009. Tracks for visualization on genome browers were generated using the BEDTools 'genomecov' utility and the UCSC bedGraphToBigWig utility (Kent, et al, Bioinformatics 26, 2204-2207, (2010); Quinlan, A. R. & Hall, I. M. Bioinformatics 26, 841-842, (2010)).
Ab initio assembly was performed using Cufflinks (2.0.2) with multi-read correction
Enabled (Trapnell, C. et al. Nature biotechnology 28, 511-515, (2010)). Gene features with the ribosomal RNA biotype 'rRNA' were added to a mask file for use with the—mask-file option in Cufflinks.
Overview of transcriptome reconstruction
To merge ab initio assembled transcript fragments (transfrags) into a consensus transcriptome a bioinformatics method that (1) classifies and filters sources of background noise in individual libraries and (2) reassembles transfrags weighted by their expression levels from multiple libraries into a consensus transcriptome was utiized.
Quality control for ab initio assembled transcripts
Ab initio assembly yielded 312,883,292 transcript fragments (transfrags) across all libraries average of 46,810 transfrags per library). Alignment artifacts and poorly assembled transcripts were controlled for by clipping very short first or last exons (< 15bp) and excluding short transfrags (< 250bp). These thresholds filtered out an average of 2.0% of the transfrags from each library, but in rare cases up to 67% of all transfrags in a library were excluded (Fig. 4a). After implementing these measures, 304,397,840 transfrags (97.2% of input) were maintained.
Assessment of genomic DNA and incompletely processed RNA levels
RNA sequencing experiments that isolate poly-adenylated RNA from whole cells inadvertently capture variable amounts of incompletely processed RNA and genomic DNA4. These noise sequences manifest within ab initio transcript assemblies as intron retentions, mono-exonic intronic transfrags in the sense orientation, and relatively lowly expressed transfrags dispersed throughout intergenic regions (Cabili, M. N. et al. Genes & development 25, 1915-1927, (2011)). Thus, background noise complicates the correct assembly of mono-exonic transcripts, intronic transcripts, or both. To characterize noise, the total unannotated sense-oriented intronic (intronic- like) transfrag population was used as a surrogate measure of both genomic and incompletely processed RNA levels, and the unannotated intergenic or antisense-oriented (intergenic-like) transfrag population as a surrogate measure of only genomic DNA levels. Comparing the transfrags in each category across all 6,503 libraries revealed significant variability in both the number and abundance of transfrags corresponding to noise (Fig. 6b). On average, intergenic-like transfrags constituted 8.6% of all transfrags (min: 0.65%, max: 43%), but only 0.88% of total FPKM per library (min: 0.16%, max: 16.8%). Intronic-like transfrags constituted 17% (min: 0.56%, max: 64%) of all transfrags and 2.0% (min: 0.18%, max: 54%) of total FPKM per library. These results implicate genomic DNA contamination and incompletely processed RNA as approximately equal contributors to total noise levels; however, these two sources of noise were not necessarily correlated.
Furthermore, individual libraries contain variable amounts of incompletely processed RNA and genomic DNA contamination. Thus, a filtering strategy that discriminated true unannotated transcription from background noise in a library-specific manner was utilized.
Filtering genomic DNA contamination artifacts from ab initio assemblies
To discriminate genomic DNA contamination from robust transcription a classification method that utilizes both relative transcript abundance and recurrence across independent biological samples was developed. The method requires a known transcript catalogue (Ensembl version 69) to determine the annotation status of ab initio transfrags (Flicek, P. et al. Ensembl 2014. Nucleic acids research 42, D749-755, (2014)). Transfrags that overlapped known transcripts in the sense orientation were denoted "annotated", and the remaining transfrags were categorized as either "Sense Intronic" or "Antisense / Intergenic" based on their relationship to annotated transcripts (Fig. 6c,d). Relative abundance was determined by using the empirical distribution of FPKM values to converting transcript FPKM values into quantiles. Recurrence levels were first computed per base by counting independent biological samples with evidence of transcription (replicates of identical cell lines or tumor tissues from the same patient were not counted towards recurrence). A single recurrence value was then computed for each transfrag by averaging the recurrence values of all bases of the transfrag. After computing relative abundance and recurrence for all transfrags, a classifier was trained to discriminate annotated from unannotated transfrags as a surrogate for classifying true transcription from background noise. Specifically, bivariate kernel density estimates were converted using the abundance-recurrence axes separately for annotated and unannotated transfrags. These densities were mapped onto a square grid (50 x 50). The annotated density was then divided by the unannotated density at each grid point after adding a nominal value to avoid floating point overflow errors. This resulted in a new grid containing likelihood ratios for annotated versus unannotated transfrags along the abundance-recurrence axes. To account for the total noise present in the library, the likelihood estimates were weighted by the relative ratio of unannotated versus annotated transfrags in the library being classified. This weight equaled the ratio of the fraction of known to unannotated transcripts in a library divided by the ratio of the medians of these fractions in all libraries. Finally, for each transfrag in an ab initio assembly, the weighted log- likelihood of the transfrag being annotated was calculated by linearly interpolating the transfrag abundance and recurrence onto the grid. For each library, a likelihood ratio cutoff was calculated by optimizing the balanced accuracy (average of sensitivity and specificity) of the classifier performance (Fig. 6e). Transfrags with likelihood below this cutoff were labeled 'background' and the remainder 'expressed'. Results from individual libraries were then concatenated to produce separate background and expressed transfrag catalogues as output. Transcripts classified as background noise were discarded and meta-assembly was carried out on the expressed fraction. To assess the sensitivity of the classification method, the filtering approach was calculated after leaving out 10% of annotated transfrags as 'test' data. The ability to detect these genes was then assessed using likelihood cutoffs determined without the test data included (Fig. lOf).
Transcriptome meta-assembly
A meta-assembly algorithm that produces isoforms from splicing partem graphs after pruning sources of incompletely processed RNA that manifest as intron retentions and inappropriately long exons is provided. Studies of alternative splicing have revealed a tightly controlled system where often only a small number of possible isoforms is observed from loci with innumerable splicing possibilities (Pickrell, J. K. et al. Nature 464, 768-772, (2010); Barash, Y. et al. Nature 465, 53-59, (2010)). To incorporate these biological observations, a greedy dynamic programming approach that reports the most highly abundant transcripts and discards minor isoforms was used.
To begin, directed acyclic splicing graphs where nodes in the graph reflect contiguous exonic regions and edges correspond to splicing possibilities were generated (Fig. 7a). Nodes in the splicing graph are then pruned according to several criteria. First, low scoring ends in the graph that correspond to extraneously long exons or overhanging exons that extend into introns are removed. Second, nodes within introns are trimmed when their scores are less than a fraction of neighboring exons. Weakly connected components of the pruned splicing graphs are then extracted and processed independently.
A splicing graph encompasses the milieu of possible isoforms that could be transcribed. Enumerating all possible paths through splicing graphs is impractical; many graphs have millions of paths of which only minute fractions are observed in vivo. The initial input transfrags provide partial paths through the splicing graph and also indicate which parts of the graph are more abundant. The approach described herein incorporates this partial path information by building a splicing pattern graph that subsumes the original splice graph (Fig. 7b). The splicing partem graph is a type of De Bruijn graph where each node represents a contiguous path of length k through the splice graph, and edges connect paths with k-1 nodes in common. As k increases so does the amount of correlative path information retained in the graph at the cost of losing short transfrags with length less than k. Each node in the graph carries a weight equal to the summed weights from all transcripts that share the node. Thus for each splice graph the partial path length k is optimized to maximize the number of nodes in the path graph with the constraint that the summed node weights of transfrags with path length greater than or equal to k is above a userspecified fraction of the total score of all transfrags. After the path graph has been constructed, every partial path transfrag is extended into a full-length transcript by transmitting the transfrag' s weight along incoming and outgoing edges. This weight is allocated proportionally at nodes with multiple incoming or outgoing edges. This approach effectively extends all partial transcript fragments into full-length transcripts and assures that the sum of incoming and outgoing node weights at equivalent. Finally, a set of isoforms is predicted from the graph using a greedy algorithm. The algorithm finds and reports the highest abundance transcript by
traversing the graph using dynamic programming. The weight of the transcript equals the minimum weight of all nodes in the path. The transcript weight is then subtracted from every node in the path and the dynamic programming procedure is repeated. Suboptimal transcripts are enumerated until a path weight falls below a fraction of the highest weighted transcript (e.g. the major isoform). The total number of isoforms produced from each gene can also be explicitly constrained. The meta- assembled isoforms are then reported in GTF and/or BED format. A genome track with summed node weights can optionally be reported in BedGraph format as well.
AssemblyLine was developed as a software package written in Python and R to
(1) characterize and filter sources of background noise in RNA-Seq assemblies and (2) perform meta-assembly to coalesce large-scale RNA-Seq datasets. AssemblyLine accepts as input a set GTF files containing transfrags assembled from individual libraries. Transfrags of length less than 250bp were omitted from meta-assembly, and the remaining transfrags were labeled as 'annotated' or 'unannotated' relative to a reference GTF file (GENCODE version 16). An ab initio transfrag was considered 'annotated' if its exons overlapping any reference transcript exons on the identical strand. A recurrence score for each ab initio transfrag was computed as the average number of samples (replicate libraries from a single cell line or tissue were considered a single sample) per nucleotide with same-stranded transcription.
Classification and filtering of 'background' and 'expressed' transfrags was performed by modeling the abundance (FPKM) and recurrence of 'annotated' and 'unannotated' transcripts using bivariate kernel density estimation on a square grid (grid size 50x50, bandwidth determined by Silverman's rule of thumb). A grid of likelihood ratios was derived from the 'annotated' and 'unannotated' grids by element-wise division at each grid point. The probability of each transfrag being 'annotated' was then determined by linearly interpolation onto this grid, and this probability was used as a surrogate measure for the probability that a transcript represented background noise. A likelihood ratio of less than or equal to one was used as a cutoff for filtering 'background' transcripts.
Filtered transcripts were subjected to the AssemblyLine meta-assembly algorithm. To limit transcript output for complex loci, isoforms with abundance less than 10% of the major transcript isoform were excluded {—fraction-major -isoform 0.10), a maximum of 20 isoforms were allowed for each gene (—maxpaths 20). During splicing pattern graph creation an optimal De Bruijn graph parameter k was determine to maximize the number of graph nodes. A maximum value of k was limited to 20 to improve the computational tractability of the optimization approach (—kmax 20). The output of meta-assembly was a GTF-formatted file as well as BED and BEDGraph-formatted files (— gtf-bed—bedgraph).
Merging of meta-assemblies
To merge meta-assemblies from 18 cohorts, the Cuffmerge tool (Trapnell, C. et al. Nature protocols 7, 562-578, (2012)), which produced a final transcriptome GTF file, was used.
Comparisons of MiTranscriptome with reference catalogs
The exons, splice sites, and splicing patterns of all assembled transcripts were compared to
RefSeq, UCSC, GENCODE (version 19), and the merged union of all three reference catalogs using custom python scripts. Sensitivity and precision values were computed using the number of shared strand-specific transcribed bases, introns, and splicing patterns. Precision was also computed for the subset of ab initio transcripts that overlapped any part of a reference transcript.
Transcripts that overlapped a reference transcript on the same strand were designated annotated. When an ab initio transcript matched multiple reference transcripts, a best match was chosen using the following criteria: (1) matching splicing pattern, (2) fraction of shared introns, and (3) fraction of shared transcribed bases. The biotype (protein, read-through, pseudogene, or lncRNA) for annotated transcripts was imputed from the best matching reference transcript. Annotated lncRNAs and unannotated transcripts were reclassified as either lncRNAs or TUCPs.
Prediction of transcripts of unknown coding potential (TUCP)
Coding potential as predicted by integrating two sources of evidence: (1) predictions from the alignment-free Coding Potential Assessment Tool (CPAT) (Wang, L. et al. Nucleic acids research 41, e74, (2013)) and (2) searches for Pfam 27.0 matches (Finn, R. D. et al. Nucleic acids research 42, D222-230, (2014)). CPAT determines the coding probability of transcript sequences using a logistic regression model built from ORF size, Fickett TESTCODE statistic55, and hexamer usage bias. A CPAT probability cutoff was chosen by repeatedly randomly sampling 100,000 each of putative non- coding and protein-coding transcripts and optimizing on the balanced accuracy (average of sensitivity and specificity) metric (Fig. 9b,c). The average area-under-the-curve (AUC) across 100 iterations was 0.9310 (minimum 0.9302, maximum 0.9320), and the average optimal probability cutoff was 0.5242 (minimum 0.5090, maximum 0.5482). This cutoff value achieved accurate discrimination of lncRNAs and protein-coding genes (sensitivity: 0.84, specificity: 0.95, FDR:
0.076). Of the putative non-coding transcripts 9,903 (5.3%) exceeded the CPAT cutoff and met the criteria for TUCP.
As additional evidence of coding potential, all transcripts were scanned for Pfam A or B domains across the three translated reading frames for stranded transcripts and six frames for monoexonic transcripts of unknown strand. To control for false positives, non-transcribed intergenic regions were scanned in the same manner. 3,781,935 hits to 12,430 unique Pfam domains in transcribed regions were observed compared with 1,774,937 hits to 1,277 unique domains in non-
transcribed intergenic space. The occurrences of each Pfam domain in transcribed versus non- transcribed regions were compared using Fisher's Exact Test and 750 domains with an odds ratio of less than 10.0 or p-value greater than 0.05 as likely artifacts were flagged (Fig. 9d). The remaining 11,726 Pfam domains were considered valid. This procedure filtered 2,972,629 artifact hits and retained 809,306 valid hits. Putative non-coding transcripts harbored only 4,674 (0.40%) of the valid Pfam domains.
The presence of Pfam domains provided strong support for CPAT coding predictions. The presence or absence of a Pfam domain stratified transcripts by the three features modeled by CPAT as well as overall coding probability (Fig. 9e). Transcripts possessing Pfam domains were much more likely to be predicted positive by CPAT than those lacking a Pfam domain (p-value < 2.2e-16, odds ratio=90.3, Fisher's Exact Test). Given the complementary aspects of Pfam domain and CPAT prediction, putative non-coding transcripts with either a Pfam domain or a positive CPAT prediction as TUCP were designed. In total 11,603 uncharacterized transcripts were flagged as TUCPs, including 5,248 transcripts previously annotated as IncRNAs. There were 2,729
uncharacterized transcripts with at least one Pfam domain, including 1,700 that did meet the CPAT criteria. By contrast, 8,874 CPAT positive transcripts lacked a valid Pfam domain. Transcripts predicted by CPAT that also harbored valid Pfam domains had longer ORFs, higher hexamer scores, and higher Fickett TESTCODE scores than other TUCPs, indicating that the Pfam and CPAT calls may be complementary (Fig. 9f-h).
Coding Potential Assessment Tool (CPAT) version 1.2.1 was performed with default parameters and used the human hexamer table and logit model (Wang, L. et al. Nucleic acids research 41, e74, (2013)). Results were scanned for Pfam 27.0 (March 2013) A and B hits using the pfam_scan.pl utility built on HMMER 3.1b (Eddy, S. R. PLoS computational biology 7, el002195, (2011); Finn, R. D. et al. Nucleic acids research 42, D222-230, (2014)). Receiver operating characteristic (ROC) analysis was performed using the ROCR package (Sing, et al, Bioinformatics 21, 3940-3941, (2005)).
Proteomics analysis
Thermo .raw files were obtained from the PRIDE database. Adul t_Kidney_Ge l_Elite_55, Adul t_Liver_Gel_Elite_56, Adul t_Pancreas_Gel_Elite_60, Adult_Rectum_Gel_Elite_63,
Adul t_Urinarybladder_Gel_Elite_40, Fetal_Brain_Gel_Velos_16, Adul t_Lung_Gel_Elite_56, and Adul t_Prostate_Gel_Elite_62. The Thermo .raw fiels were transformed into mzXML using
MSConverter and interrogated against human UniProt database V.15.11 using X! tandem search engine. The database was concatenated with all possible open reading frames longer than 7 amino acids from IncRNA database and with reversed sequences for determination of FDR. The X! Tandem
search parameters were: fully tryptic cleavage, parent mass error 5 ppm, fragment mass error 0.5 Da, 2 allowed missed cleavages. Fixed modifications: Cys carbamidomethylation. Variable
modifications: Met oxidation. X! Tandem output files were processed by PeptideProphet and ProteinProphet and for final output the data was filtered at peptide probability 0.5 and protein probability 0.9 to ensure protein FDR < 1%.
Confidence scoring system
After assembly of the MiTranscriptome, transcripts were subjected to an additional confidence evaluation. IncRNAs in the MiTranscriptome were categorized into tiers based on their annotation status and the degree of matching of splice junctions to the reference annotation. Tier 1 transcripts are all annotated and tier 2 transcripts are unannotated. An empirical cumulative distribution function was developed by profiling the second highest expression value (across all 6,503 samples) for each tier 1 transcript. The second highest value was used to control for outlier expression. The second highest expression value for each tier 2 transcript was then fed into the distribution function to produce the confidence score.
Validation of IncRNA transcript by qRT-PCR
150 IncRNAs with at least 1 FPKM expression in either A549, LNCaP, or MCF7 cells were chosed for biological validation. For each transcript, primer pairs were designed using the Primer- BLAST tool. Primer pairs with the following parameters were selected: (1) amplicon length between 80-140 bp (2) primer GC content between 35-65%, and (3) primer length greater than 20 bp. Primers were blasted against the human genome to ensure specificity to the target gene, and primers designed against multiexonic transcripts spanned exon junctions. Regions of any transcript that directly overlapped an exon on the antisense strand were avoided. Primer pairs meeting these criteria could be designed for 100 out of 150 IncRNAs (38 monoexonic and 62 multiexonic). All oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA).
RNA was isolated from A549, LNCaP and MCF7 cells in Trizol (Invitrogen) using the
RNeasy Mini Kit (Qiagen). Equal amount of RNA was converted into cDNA using random primer's and the Superscript III reverse transcription system (Invitrogen). Quantitative real-time PCR (qPCR) was performed using Power SYBR Green Mastermix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7900HT Real-Time PCR System. The housekeeping genes, CHMP2A, EMC 7, GPI, PSMB2, PSMB4, RAB7A, REEP5, SNRPD3 were used as loading controls56. Data was normalized first to housekeeping genes and then to the median value of all samples using the delta-delta Ct method and plotted as fold change over median. To ensure the specificity of the primers, 20 amplicons were further analyzed by Sanger sequencing.
Cell lines and reagents:
All cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cell lines were maintained using standard conditions. Specifically, A549 were grown in F-12K plus 10% fetal bovine serum (FBS), LNCaP in RMPI1640 (Invitrogen) plus 10% FBS and 1% penicillin-streptomycin, and MCF7 in Eagle's Minimum Essential Media (EMEM) plus 10% FBS. All of the cell lines were grown at 37°C degrees in a 5% C02 cell culture incubator. To ensure identity, cell lines were genotyped at the University of Michigan Sequencing Core using
Profiler Plus (Applied Biosystems) and compared with the short tandem repeat (STR) profiles of respective cell lines available in the STR Profile Database (ATCC). All of the cell lines were routinely tested and found to be free of Mycoplasma contamination.
Evidence for active regulation of transcriptional start sites
To conduct analysis of TSS intervals ENCODE project datasets were downloaded from the UCSC Genome Browser (Karolchik, D. et al. Nucleic acids research 42, D764-770, (2014)). For H3K4me3 analysis the Encode Project Broad Institute H3K4me3 ChlP-Seq peaks for cell lines GM12878, Hl-hESC, HeLa-S3, HepG2, HMEC, HSMM, HSMMtube, HUVEC, K562, NH-A, NHDF-Ad, NHEK, and NHLF57 were used. For RNA polymerase II analysis POL2RA binding sites from the Encode Project Uniform TFBS master file version 3 for any of the cell lines with H3k4me3 data were used (Consortium, E. P. et al. Nature 489, 57-74, (2012)). Finally, for DNase
hypersensitivity analysis the Encode Project combined UW and Duke DNasel hypersensitivity regions were downloaded as a master file from EMBL-EBI, and filtered for any of the cell lines with H3k4me3 data. Peak enrichment files (BED format) were aggregated across all cell lines.
Intervals of +/- 10 kilobases surrounding unique MiTranscriptome TSSs were generated using BEDTools 'slop' tool (Quinlan, A. R. & Hall, I. M. Bioinformatics 26, 841-842, (2010)). TSSs were filtered for expression in each cell line at RPKM>0.1.Basewise peak coverage was generated for each TSS interval using the BEDTools 'coverage' function and summarized across subsets of TSSs. Summed per-base coverage histograms were normalized by dividing by the number of expressed TSSs.
Conservation analysis
The evolutionary conservation of transcripts in the assembly was studied using two metrics: (1) the fraction of significantly conserved bases (p < 0.01, phyloP algorithm), and (2) the maximally conserved 200nt sliding window (phastCons scores averaged within each window). The former captures independently conserved elements within a transcript regardless of position, and the latter captures contiguous regions of high conservation. The 200nt sliding window size was chosen to aid in discovery of putative ultraconserved elements (Bejerano, G. et al. Science 304, 1321-1325, (2004)). As a negative control the conservation of non-transcribed regions was measured using these
metrics by randomly sampling contiguous length-matched intervals from intergenic and intronic space. Non-transcribed interval sampling was restricted to regions with valid 46-way conservation data. The fractional basewise conservation and contiguous window conservation metrics were used to nominate highly conserved and ultraconserved transcripts, respectively. In both cases cutoffs for significant transcripts were determined by controlling the rate of observing elements with similar conservation levels within non-transcribed intergenic space at a level of 0.01. For fractional basewise conservation a score of 0.0947 (9.5% of transcript bases conserved at phyloP p-value <0.01) corresponded to a false discovery rate < 0.01. At this cutoff the sensitivity for detecting protein- coding transcripts was 0.67. For contiguous sliding window conservation an average PhastCons probability of 0.9986 corresponded to a false discovery rate < 0.01. At this cutoff the sensitivity for detecting true positive ultraconserved non-coding elements downloaded from UCNEbase was 0.6926. Applying these criteria to the assembly yielded 6,034 IncRNAs (3.4%) and 541 TUCPs (4.7%) with significant basewise conservation levels. Additionally, 1 ,686 IncRNAs (0.96%) and 121 TUCPs (0.01%) harbored contiguous ultraconserved regions.
GWAS analysis
A list of GWAS SNPs was obtained from the National Human Genome Research Institute's GWAS catalog (Welter, D. et al. Nucleic acids research 42, D1001 -1006, (2014)). SNP haplotypes were excluded from the SNP overlap analysis, and a list of 1 1, 194 unique SNPs was obtained. The merged union of the RefSeq, UCSC, and GENCODE catalogs was used as a reference for comparison with MiTranscriptome.
Genomic conservation profiles generated by the phyloP (phylogenetic p-values) and
PhastCons algorithms for multiple alignments of 45 vertebrate genomes to the human genome were downloaded from the UCSC genome browser (Karolchik, D. et al. Nucleic acids research 42, D764- 770, (2014); Pollard, et al, Genome research 20, 1 10-121 , (2010); Siepel, A. et al. Genome research 15, 1034-1050, (2005)). The 'wigFix' formatted files were converting into 'bigWig' formatted files using the 'wigToBigWig' binary utility program provided by the UCSC genome browser (Karolchik et al., supra). For each transcript a vector of conservation scores for each exon was extracted using the 'bigWigToBedGraph' utility and concatenated into a single vector. Conservation metrics were then computed from these vectors.
Intersections of GWAS SNPs with transcripts or exons was performed using the
BEDtools 'intersect' tool, with the '-split' option invoked for quantification of exonic
Overlap (4. The number of GWAS SNPs overlapping the entire assembly and individual transcript categories (lncRNA,TUCP, pseudogene, protein-coding, and read-through) was determined by BEDTools 'intersect' for both the whole transcript and for exonic regions {nGWAS). Subsequently, a
set of all the SNPs from two popular SNP arrays (Illumina HumanHap550 and Affymetrix SNP6) was created, which was termed the "SNP background". The amount of SNPs from the SNP background overlapping the MiTranscriptome was calculated (nback ground), and the fraction of the number of overlapping GWAS SNPs to the number of overlapping SNPs from the SNP background (fracGWAS = nGW AS I nback ground) was then reported for each category. This fraction was also calculated using random shuffling of the MiTranscriptome and its components into noncoding regions of the genome (fracshuffle). One hundred shuffles were performed for each condition, and an odds ratio (ORGWAS = fracGWAS/ fracshuffle) was determined for each shuffle. The purpose of using fracGW ASinstead of simply using nGW AS in this analysis is to control for the possibility that during the shuffle, transcripts could be shuffled into regions not represented on SNP arrays (e.g., regions unable to possess GWAS SNPs), falsely lowering the amount of GWAS SNP overlap by the shuffle. If transcripts are shuffled into regions that are not represented by the SNP background, both nGW AS and nbackground will decrease together, with fracGWAS relatively unchanged.
Shuffling was performed using the BEDTools 'shuffle' tool. MiTranscriptome transcripts were grouped by transcription locus (e.g., regions of the genome that have contiguous transcription) prior to shuffling. Shuffling of transcript loci was performed to control for the fact that transcripts within a locus are spatially linked to one another. Shuffling without locus clustering would falsely elevate the amount of genome covered by transcripts, and subsequently elevate the number of SNPs overlapping the shuffled regions. A concatenation of the UCSC hgl9 gaps file and the
MiTranscriptome proti en-coding transcripts was used as an exclusion file for these shuffles.
As a negative control, the entire above analysis was repeated using and equal number randomly selected SNPs (chosen from the Illumina HumanHap550 and Affymetrix SNP6 background) in place of the GWAS SNPs. The significance of enrichment for GWAS SNPs versus random SNPs was measured across identical shuffles of the transcript loci using paired Student's t- tests comparing the set of odds ratios for all shuffles. Similar analysis to determination of compendia enrichment was performed to identify enrichment of novel intergenic IncRNAs and TUCPs. The intergenic space was defined as all regions not covered by the merged reference. For this analysis, the shuffles were performed into the intergenic space, instead of all non-coding space. The exclusion file used by BEDtools 'shuffle' was a concatenation of the UCSC gaps file and the merged reference. Transcript expression estimation
Expression levels (FPKM) of the transcripts in the assembly were determined using Cufflinks (version 2.02 and 2.1.1)60. Normalized abundance estimates (FPKM) were computed for all MiTranscriptome transcripts, converted into approximate fragment count values, and aggregated into
a matrix of expression data (Fig. 3a). Library size factors for expression normalization were computed by applying the geometric normalization method described by Anders and Huber (Genome biology 11, Rl 06 (2010)).
The transcript abundances for all transcripts in the MiTranscriptome assembly were estimated using Cufflinks version 2.1.15 with the following parameters : ' ~max-fragmultihits=
, '--no-effective-length-correction', '--max-bundle-length 5000000', '--maxbundle- frags
20000000'. To convert normalized transcript abundance estimates (FPKM) to approximate fragment count values each FPKM is multiplied by the transcript length (in kilobases) and by the "Map Mass" value (divided by 1.0e6) reported in the Cufflinks log files. By some reverse engineering and assistance from the seqanswers online forum (seqanswers.com), it was determined that this factor was utilized in the normalization process. Abundance estimation for 28 libraries failed for technical reasons (corrupt BAM files) and these libraries were discarded from the expression analysis.
Expression estimation for 2,246 transcripts yielded errors and/or zero-valued counts and were discarded.
Transcript expression enrichment analysis
To analyze differential expression of transcripts relative to sample phenotypes a method called Sample Set Enrichment Analysis (SSEA) was developed. The source code for this software is available online. The method adapts the weighted Kolmorgorov-Smirnoff (KS) tests proposed by Gene Set Enrichment Analysis (GSEA). In contrast to GSEA, which tests for associations with gene sets, SSEA tests for associations between individual gene expression observations (which could be transcript or gene expression) and sample sets. Thus, SSEA is analogous to performing GSEA on a 'transposed' input dataset. However, SSEA incorporates important features not provided by GSEA:
(1) methodology for non-parametric analysis of discrete count data (e.g. RNA-Seq count datasets),
(2) engineering improvements to enable analysis of big datasets (here, a matrix of 381,731 rows and 6,475 columns was analyzed using less than 1 Gb of RAM), and (3) parallelization of the algorithm for use in high performance computing environments.
Differential expression testing was performed using the Sample Set Enrichment Analysis method developed as part of this study. SSEA was performed with 100 iterations of count resampling and 1,000 null permutations for each transcript (~resampling-iterations=100, ~perms=1000). These parameters yielded a minimum FDR resolution of approximately le-7 for all sample sets. Weights for the KS-test were log(x + l)-transformed normalized count values (~weight-hit=log,—weight- miss=log, ~weightparam= 1).
KS-tests using normalized count data vectors as weights.
To convert count values into weights for a single KS-test the following steps are performed: (1) raw count values are normalized by library-specific size factors, (2) normalized count values are "resampled" from a Poisson distribution (lambda equals the observed count value) to mimic the effect of technical replication, and (3) random Poisson noise (by default, lambda equals 1) is added to the normalized, resampled count values to destabilize zero-valued counts and break ties. A power transform (exponential or logarithmic) is then applied to the weights (by default, a logtransformation is applied after incrementing normalized count values by 1). The choice of power transformation influences the relative importance of precision versus recall during enrichment testing. For example, users aiming to discover genes new in molecular subtypes of a disease would prioritize precision over sensitivity, whereas a user aiming to discover ideal biomarkers may value sensitivity over precision. Following count data normalization and power transformation, SSEA performs the weighted KS-test procedure described in GSEA28. The resulting enrichment score (ES) statistic describes the strength of association between the weights and the sample set.
To control for random sampling bias in count values (e.g. "shot noise") SSEA performs repeated enrichment tests using resampled count values to mimic observations from technical replicates and uses the median enrichment score (by default, 100 tests are performed). The basis for Poisson resampling as a legitimate model for technical replication was established by Marioni et al.62 To test for significance, SSEA performs enrichment tests using randomly shuffled sample labels to derive a set of null enrichment scores with the same sign as the observed score (by default, 1000 null enrichment scores are computed). The nominal p value reported is the relative rank of the observed enrichment score within the null enrichment scores. To control for multiple hypothesis testing, SSEA maintains the null normalized enrichment score (NES) distributions for all transcripts in a sample set, and uses the null NES distribution to compute FDR q values in the same manner as proposed by Subramanian et al. (Proceedings of the National Academy of Sciences of the United States of America 102, 15545-15550, (2005)).
Benchmarking SSEA performance using microarray gene signatures
Gene signatures for the top 1% of overexpressed and underexpressed genes from three prostate cancer (Grasso, C. S. et al. Nature 487, 239-243, (2012); Taylor, B. S. et al. Cancer cell 18, 11-22, (2010); Yu, Y. P. et al. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 22, 2790-2799, (2004)) and three breast cancer (Cancer Genome Atlas, N. Nature 490, 61-70, (2012); Curtis, C. et al. Nature 486, 346-352, (2012); Gluck, S. et al. Breast cancer research and treatment 132, 781-791, (2012).) microarray studies were obtained using Oncomine (Rhodes, D. R. et al. Neoplasia 9, 166-180 (2007).). The top 1% gene signatures as detected by SSEA in the MiTranscriptome breast and prostate cohorts were determined using
prostate cancer versus normal and breast cancer versus normal sample sets (Fig. 3a). Given that the MiTranscriptome was produced from an ab initio assembly, transcript identity was assigned to the annotated reference gene with the greatest degree of concordance, where degree of splicing agreement was prioritized over degree of exonic same-stranded overlap. The most-enriched isoform for each gene was used to produce a gene signature.
Degree of overlap for all combinations of the 16 gene sets tested (3 published breast up- regulated sets, 3 published breast down-regulated sets, 3 published prostate up-regulated sets, 3 published prostate down-regulated sets, 1 SSEA-determined prostate up-regulated set, 1 SSEA- determined prostate down-regulated set, 1 SSEA-determined breast up-regulated and 1 SSEA- determined breast down-regulated set) was determined by calculating an odds ratio and performing a Fisher's exact test for each gene set pair. Each comparison was restricted to the set of genes assessed by both profiling platforms. Microarray chip annotation files were downloaded from the Molecular Signatures Database (MSigDB) web site (Subramanian, A. et al, supra). The set of all annotated genes (relative to RefSeq, UCSC, and GENCODE) was used as the annotation file for
MiTranscriptome.
Discovery of lineage-specific and cancer -specific transcripts
To generate enrichment test data for unsupervised clustering, transcripts were ranked within each SSEA sample set by normalized enrichment score (NES) and assigned fractional ranks (e.g. a fractional rank of 0.95 implies the transcript ranked in the top 5th percentile of all transcripts in the sample set). Only significant results (FDR < le-7 for lineage analysis and FDR < le-3 for cancer versus normal analysis) were used. Unsupervised clustering was performed using Pearson correlation of log-transformed fractional ranks as a distance metric and Ward's method. Transcripts that were significantly associated with multiple sample sets were grouped with the most strongly associated sample set. Heatmaps were produced using the 'heatmap.2' function from the 'gplots' package in R. Guilt-by-association GSEA analysis
For each cancer and/or lineage associated lncRNA, expression levels of the target lncRNA were correlated to the expression of all protein-coding genes across all samples in the associated tissue cohort. For cancer cohorts (e.g. breast, prostate), correlations were performed (Spearman) using only the cancer samples (normal samples were excluded). To account for multiple isoforms of eachThe protein-coding genes were then ranked by the Rho value, and used in a weighted, pre- ranked GSEA analysis against a collection of cancer associated gene sets from MSigDB. Significant associations were determined for any gene set having an FWER p-value below 0.001.
Results
An expanded landscape of human transcription
The spectrum of human transcriptional diversity was investigated by curating 7,256 poly-A+ RNA-Seq libraries from 25 independent studies, including 5,847 from TCGA, 928 from the Michigan Center for Translational Pathology (MCTP), 67 libraries from the Encyclopedia of DNA Elements (ENCODE), and 414 samples from other public datasets (Fig. 5a). An automated transcriptome assembly pipeline was developed and employed to process the raw sequencing datasets into ab initio transcriptome assemblies (Fig. 5b). This bioinformatics pipeline utilized approximately 1,870 core-months (average 0.26 core-months per library) on high-performance computing environments.
Collectively the RNA-Seq data constituted 493 billion fragments; individual libraries averaged 67.9M total fragments and 55.5M successful alignments to human chromosomes. On average 86% of aligned bases from individual libraries corresponded to annotated RefSeq exons, while the remaining 14% fell within introns or intergenic spacel5. Coarse quality control measures were used to account for variations in sequencing throughput, run quality, and RNA content by removing 753 libraries with (1) fewer than 20 million total fragments, (2) fewer than
20 million total aligned reads, (3) read length less than 48bp, or (4) fewer than 50% of aligned bases corresponding to RefSeq genes (Figs. 5c,d). After coarse filtration, approximately 391 billion aligned fragments (43.69 terabases of sequence) were identified for subsequent analysis. The set of 6,503 libraries passing quality control filters included 6,280 datasets from human tissues and 223 samples from cell lines. Of the tissue libraries, 5,298 originated from primary tumor specimens, 281 from metastases, and 701 from normal or benign adjacent tissues (Figs. 5e). This set of samples is referred to as the MiTranscriptome compendium.
Upon processing the MiTranscriptome libraries, ab initio transcriptome were obtained reconstructions from 6,503 individual tumors, normal tissues, or cell lines. A computational methodology was developed to coalesce individual transcriptomes into a consensus transcriptome, a procedure known as 'meta-assembly'. Unlike previous methods for meta-assembly of expressed sequence tag (EST) data or small numbers of RNA-Seq experiments, the meta-assembly utilized in this study addressed computational and scalability challenges stemming from the magnitude of this study (Haas, B. J. et al. Nucleic acids research 31, 5654-5666 (2003); Trapnell, C. et al. Nature protocols 7, 562-578, (2012)).
To permit sensitive detection of lineage-specific transcription the libraries were partitioned into 18 cohorts by organ system (Fig. la), performed filtering and meta-assembly separately for each cohort, and re-merged the cohorts (Fig. lb). The individual ab initio assemblies collectively totaled -312M transcript predictions (transfrags) across all libraries. To perform filtering, short transfrags (<250bp) and clipped short flanking exons (<15bp) were removed, leaving -304M transfrags (Fig.
6a). Whereas levels of annotated transfrags were relatively constant, fractions of unannotated intragenic and intergenic transcripts varied considerably across libraries (Fig. 6b). Almost one-third of all transfrags were unannotated (29.3%, or 89M), including 86.2M mono-exonic and 2.8M multiexonic transfrags. Two sources of background noise in RNA-Seq experiments that could give rise to unannotated mono-exonic transfrags are incompletely processed RNA and genomic DNA contamination (Fig. 6c). To minimize this noise, a conservative filtering scheme was used (Fig. 6d). 60M mono-exonic transfrags within introns that could have arisen from incompletely processed RNA were discarded. A machine learning method was developed to discriminate recurrent antisense and intergenic transcription from possible genomic DNA contamination. The approach models the empirical distributions of relative transcript abundance and recurrence (number of independent samples in which the transcript was observed) to determine optimal library-specific thresholds for distinguishing annotated from unannotated transcription. The classifier achieved remarkable performance (average AUC of 0.89, range 0.77-0.96) and displayed no bias for cancer versus normal samples (Fig. 6e). Moreover, the classifier recovered test transcripts left out of the training process with 80% mean sensitivity (range 0.64-0.95, Fig. 6f). Ultimately 3.2M of the 86.2M (3.7%) mono- exonic intergenic or antisense transfrags were retained for a total of 6.0M unannotated transfrags (6.75% of the original 89M). The filtered collection of 221M annotated and unannotated transfrags was subjected to meta-assembly. The meta-assembly algorithm first collapses transfrags into a splice graph and utilizes transcript abundance information to prune intron-retentions and trim long first or last exons (Fig. 7a). Furthermore, the algorithm integrates splicing pattern information by constructing a splicing pattern graph and traverses the graph using a greedy dynamic programming algorithm to generate full-length transcript predictions (Fig. 7b). For example, meta-assembly of 7,471 transfrags in the chromosome 12 locus containing HOTAIR and HOXC11 produced just 17 transcripts, including transcripts that accurately matched annotated HOTAIR and HOXCl 1 isoforms (Fig. 7c). After merging meta-assemblies from 18 cohorts, a consensus set of 384,066 predicted transcripts designated as the MiTranscriptome assembly was identified.
To begin characterizing the assembly, comparisons with reference catalogs from RefSeq (Dec, 2013) (Pruitt, K. D. et al. Nucleic acids research 42, D756-763, (2014)), UCSC (Dec, 2013) (Karolchik, D. et al, supra), and GENCODE (Release 19) (Harrow, J. et al. Genome
research 22, 1760-1774, (2012)). (Fig. lc) were performed. In particular, increases in numbers of exons, splice sites, transcripts, and genes of 29%, 52%, 95%, and 57%, respectively, were observed relative to GENCODE, the most expansive of the three reference catalogs. To understand the source of the increases, the assembly was overlapped with a merged union of the three reference catalogs
and the fraction of unannotated versus annotated transcripts were delineated for each cohort (Fig. 8a).
Analysis of the assemblies on the cohort level reveals that the majority of transcripts assembled within each lineage cohort overlapped annotated genes (range 62-88%, mean 75%).
However, the fraction of annotated genes within the entire MiTranscriptome (a merger of the 18 individual cohorts) was just 46%, indicating the presence of much unannotated transcription unique to specific lineages. The sensitivity and precision for detecting annotated nucleotides, splice sites, and splicing patterns in the three reference catalogs and intergenic IncRNA predictions from the previous cataloguing study by Cabili et al. (Cabili, M. N. et al. Genes & development 25, 1915-1927, (2011)) were quantitated (Fig. 8b,c). The MiTranscriptome assembly was very sensitive to detection of annotated transcribed bases and splice sites. For example, the MiTranscriptome detected 94% and 93% of annotated RefSeq bases and splice sites, respectively. Detection of precise splicing patterns remains an ongoing challenge for in silico transcriptome reconstruction methods (Steijger, T. et al. Nature methods 10, 1177-1184, (2013)).
Coding potential assessment of long RNA transcripts
To facilitate further study of the assembly, transcripts were classified into one of five categories: (1) Protein-coding, (2) Read-through (implying a transcript overlapped multiple separate annotated genes), (3) Pseudogene, (4) IncRNA, and (5) Transcript of Unknown Coding Potential (TUCP) (Fig. 9a). The TUCP classification was originally described by Cabili et al. (supra) and pertains to long RNAs with features indicative of coding potential but not already annotated as protein coding. The ability to predict coding potential in silico using sequence features alone has important implications for ab initio transcript annotation studies. Here, TUCPs were predicted by incorporating two methods: (1) predictions from the Coding Potential Assessment Tool (CPAT) (Wang, L. et al. Nucleic acids research 41, e74, (2013)), which analyzes the sequence features of transcript open reading frames (ORFs), and (2) presence of a known Pfam domain (Finn, R. D. et al. Nucleic acids research 42, D222-230, (2014)) within a transcript ORF (Fig. 9b-h). Over sixty percent of all MiTranscriptome genes were classified as either IncRNAs or TUCPs (59% lncRNAs, 3.5% TUCPs, Fig. 2a). The majority of IncRNAs and TUCPs were unannotated relative to RefSeq, UCSC, and GENCODE genes (79% and 66%, respectively) and located within intergenic regions (72% and 60%, respectively) (Fig. 2b). 5,248 transcripts overlapping annotated IncRNAs were flagged as TUCPs, indicating that previous annotation attempts may have identified incomplete ostensibly noncoding fragments that may actually comprise transcripts possessing robust ORFs. For example, in a chromosome 16 intergenic locus, transcripts harboring a 418 amino acid ORF spanning 29 exons that overlapped three independent genes annotated by GENCODE as IncRNAs (LINC00514, LA16c-
380H5.3, LA16c-380H5.4) were identified, indicating that the annotated GENCODE IncRNAs may be incomplete partial annotations of a larger protein-coding gene (Fig. 2c).
To further investigate the coding potential of these TUCP transcripts, a proteomics analysis was performed to search for reported peptides that may map to ORFs in the TUCPs. Recent proteomics studies have produced the most comprehensive analysis of the human proteome to date (Kim, M. S. et al. Nature 509, 575-581, (2014)). Using these data, it was assessed whether any novel, uniquely mapping peptides map to an ORF in any of the TUCP transcripts. Many novel and uniquely mapping peptides in various tissue types mapped to ORFs in the TUCP transcripts, with a total of 268 TUCP genes possessing matching peptides. These and other TUCP predictions exemplify the potential for MiTranscriptome to enhance reference transcript catalogs.
Characterization and validation of long RNA transcripts
LncRNA and TUCP genes tended to have fewer exons than read-through or protein coding genes, but appreciable alternative splicing was observed for all classes of transcripts (Cabili et al, supra; Derrien, T. et al. Genome research 22, 1775-1789, (2012).) (Fig. 10a). Furthermore, it was observed that IncRNAs and TUCPs were expressed at lower levels than read-through or protein- coding transcripts, which is also consistent with previous studies (Prensner, J. R. et al. Nature biotechnology 29, 742-749, (2011)); Cabili et al, supra; Derrien et al, supra; Guttman, M. et al. Nature biotechnology 28, 503-510, (2010)) (Fig. 2d).
To characterize transcription start sites (TSS), intervals surrounding TSSs with ENCODE histone 3 lysine 4 trimethylation (H3K4me3) ChlP-Seq, RNA polymerase II (PolII) binding sites, and DNase hypersensitivity data from 13 cell lines were compared. To control for expression, binding was only assessed for transcripts expressed in the cell lines being assayed, filtered TSSs for expression before intersection at a level of FPKM>0.1. LncRNA and TUCP promoters were enriched for these marks relative to randomly shuffled control regions, with maximal enrichment at the TSS (Fig. 2e-g). Enrichment was lower for IncRNA and TUCP promoters than for protein-coding genes, but much more enriched than pseudogenes, which may reflect their overall lower expression levels. These chromatin modification and polymerase binding data indicate that the assembled IncRNA and TUCP transcripts possess actively regulated promoters.
During assembly of the MiTranscriptome, a first-pass filtering of low-confidence transfrags was performed via a machine-learning algorithm built using the expression level and recurrence of the transfrag (Fig. 6d). Millions of transfrags were removed at this step, and the resultant
MiTranscriptome contains only transcripts that have met this first-pass confidence evaluation. To further stratify the confidence transcripts, a confidence score (CS) system was developed. IncRNAs were classified into two tiers based on their annotation status and the matching of splice junctions,
and a cumulative distribution function was built using the expression levels for the annotated IncRNAs (tier 1). The expression level of each unannotated lncRNA (tier 2) was then fed into the cumulative distribution function to calculate a CS for each lncRNA (Fig. 10b). The CS profile of the tier 1 and tier 2 transcripts was largely similar, with a slight enrichment in low confidence transcripts among the unannotated transcripts (e.g. 32% of unannotated IncRNAs have CSs lower than the bottom 12.5th percentile of annotated IncRNAs). This phenomenon, however, can be explained by a discovery bias given that the confidence metric is expression based. To further strengthen confidence in the assembly transcripts, the predicted lncRNA expression was validated by qRT-PCR. qPCR primers were developed for 100 candidate IncRNAs. Three cell lines were selected representing lung cancer, prostate cancer and breast cancer (A549, LNCaP, MCF7, respectively), and IncRNAs with expression of at least 1 FPKM by RNAseq in at least one of the cell lines were selected for validation (38 monoexonic, 62 polyexonic). Given that genomic contamination can produce spurious monoexonic reads during assembly, an absence of reverse transcriptase (-RT) was used as a control for this study. Of the 100 IncRNAs tested, 95 had significantly higher expression with reverse transcriptase when compared to -RT (Student's t-test, p-value < 0.05) in cell lines for which expression was expected via RNA-Seq (>1 FPKM) (Fig. 11). DSCAM-AS1 and PC ATI 30 are two examples of IncRNAs nominated by SSEA analysis to have cancer specificity (in breast and prostate, respectively) whose cell line expression profile by qRT-PCR reflects what is expected from the tissue SSEA analysis (Fig. 12, boxed genes).
To further ensure that the amplicon was from the expected gene, twenty of the most expressed transcripts across the three cell lines (according to the qRT-PCR data) were selected and their identity confirmed by Sanger sequencing. In eighteen of the twenty cases, the sequence of the exact gene of interest was amplified (Fig. 12a,b). Additionally, the expression values identified by qRT-PCR for each cell line were correlated to the RNA-seq FPKM values in each cell line. qRT- PCR was correlated best with RNA-seq expression from the same cell line (Fig. 12c).
LncRNAs harboring conserved elements
The evolutionary conservation of IncRNAs has been a topic of ongoing conversation, with several reports indicating that IncRNAs are modestly conserved (Cabili et al, supra; Derrien et al. supra; Necsulea, A. et al. Nature 505, 635-640 (2014)). In agreement with previous reports, increases in both transcript and promoter conservation levels for IncRNAs and TUCPs relative to random control regions were observed (Fig. lOc-f). Shifts in the cumulative distributions of lncRNA and TUCP transcripts were greater for annotated transcripts relative to unannotated transcripts. This difference may reflect discovery bias favoring highly conserved genes detectable across multiple model systems. Despite observing increased conservation within the entire class of IncRNAs, the
results indicated that human IncRNA conservation may be an exceptional phenomenon rather than a general one; therefore, IncRNAs harboring higher than expected basewise conservation were selected for focused study (Fig. 2h). 3,309 IncRNA genes (5.6% of all IncRNAs) that were highly conserved relative to random intergenic regions were selected (Fig. lOe). In addition, part of the noncoding genome includes ultraconserved elements (UCE), which are stretches of DNA >200nt with nearly perfect sequence identity across multiple organisms (Bejerano, G. et al. Science 304, 1321-1325, (2004); Dimitrieva, S. & Bucher, P.
Nucleic acids research 41, D101-109, (2013)). 597 intergenic IncRNAs (1.2% of all intergenic IncRNAs) harboring UCEs were designated as Highly Conserved Long Intergenic Non-Coding RNAs (HICLINCs) to promote further study of transcribed UCEs as a class (Fig. lOh). For example, THCAT126, a previously unannotated intergenic IncRNA on chromosome 2q24, contains elements in its final exons that are conserved in nearly all vertebrates including zebrafish (Fig. 2i). Moreover, THCAT126 is expressed widely across many tissue types, and is expressed in multiple cancers, with a significant association in the thyroid cancer versus normal analysis (Fig. 2j). Highly conserved IncRNAs such as THCAT126 (and many other cancer-associated HICLINCs described below) provide an avenue for in vivo study of the role of IncRNAs in development and cancer.
LncRNAs overlapping disease-associated SNPs
To investigate the relationship of the MiTranscriptome assembly with disease-associated regions of the genome, overlap of transcripts in the assembly was compared with 11,194 unique disease associated single nucleotide polymorphisms (SNPs) from a catalog of genome-wide association studies (GWAS) (Welter, D. et al. Nucleic acids research 42, D1001-1006, (2014)). MiTranscriptome transcripts overlapped 9,770 GWAS SNPs compared to just 7,050 SNPs overlapping GENCODE, UCSC, or RefSeq transcripts. Exonic overlap was 2,586 and 1,096 GWAS SNPs for the MiTranscriptome and aggregated reference catalogs, respectively (Fig. 13a,b).
Altogether transcripts in the assembly coincided with 2,881 formerly intergenic SNPs located within 'gene deserts', and only missed 161 GWAS SNPs overlapping annotated genes. It was observed that the increased overlap with GWAS SNPs for MiTranscriptome transcripts and exons were significantly enriched for GWAS SNPs relative to random SNPs chosen from the same chip platform (paired t-test, p-value, 5.25e-135 and 1.15e-199, respectively, Fig. 2k). Moreover, unannotated intergenic IncRNAs and TUCPs were also significantly enriched for disease-associated regions, with exons more highly enriched than full-length transcripts (paired t-test, p-value, 9.90e-78 and 5.50e-50, for whole transcript and exon, respectively, Fig. 13c). These data indicate that a rigorous
reevaluation of allele-specific gene expression regulation in regions proximal to GWAS SNPs yields informative biological associations with the new IncRNA transcripts identified in this study.
Detection of cancer-associated transcription by enrichment analysis
The large-scale transcriptome reconstruction process unveiled tremendous transcriptional complexity highlighted by the presence of thousands of uncharacterized IncRNAs and TUCPs. To prioritize disease-associated and lineage-specific transcription, a nonparametric method for differential expression testing called Sample Set Enrichment Analysis (SSEA) was used. SSEA adapts the weighted Kolmorgorov-Smirnoff-like tests used by Gene Set Enrichment Analysis (GSEA) (Subramanian, A. et al. Proceedings of the National Academy of Sciences of the United States of America 102, 15545-15550, (2005)) to discover transcript expression changes associated with predefined sample sets. This method permits sensitive detection of differential expression within heterogeneous sample populations (e.g., tumor sub-types). Prior to running SSEA, isoform- level expression data for the entire MiTranscriptome assembly was re-computed and samples from the compendia were grouped into fifty sample sets. A sample set represents a single condition for evaluating differential transcript expression. The sets in the present study included various cancer types (e.g., prostate cancers versus all other MiTranscriptome samples), normal tissues or cell types, and cancer versus normal comparisons within a single tissue type (e.g., prostate cancers versus benign prostate samples) (Fig. 3a). All MiTranscriptome transcripts were tested against the fifty samples sets, and collectively, SSEA detected over two million significant associations (FDR < le-3 for cancer versus normal analyses and FDR < le-7 for lineage analyses) involving 267,726 of the 381,821 MiTranscriptome transcripts for which enrichment analysis was possible.
To validate the enrichment testing approach, its ability to rediscover known proteins up- regulated and down-regulated in prostate cancers and breast cancers was assessed by assessing the concordance between the top 1% positively and negatively enriched genes from each cancer type with cancer gene signatures obtained from the Oncomine database of microarray studies (Rhodes, D. R. et al. Neoplasia 9, 166-180 (2007); Cancer Genome Atlas, N. Nature 490, 61-70, (2012); Curtis, C. et al. Nature 486, 346-352, (2012); Gluck, S. et al. Breast cancer research and treatment 132, 781-791, (2012); Grasso, C. S. et al. Nature 487, 239-243, (2012); Taylor, B. S. et al. Cancer cell 18, 11-22, (2010); Yu, Y. P. et al. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 22, 2790-2799, (2004)). A heatmap of the odds ratios of the gene signature associations revealed striking agreement between SSEA and the other studies for both cancer types, with SSEA often demonstrating equal or better concordance to each microarray study than comparison of the microarray studies to each other (Fig. 3b). Thus, isoform-level differential expression testing from the MiTranscritpome ab initio assembly of RNA-Seq data recapitulated the results from cancer microarray gene expression studies, supporting the SSEA method as a viable tool for detection of differential expression. To further credential the enrichment testing approach, the
ability to detect positive control IncRNAs and protein-coding genes in breast cancers and prostate cancers was assesed. For example, SSEA correctly identified the oncogenic IncRNA HOTAIR7, estrogen receptor 1 (ESR1), and GATA binding protein 3 (GAT A3) as highly positively enriched in breast cancers (Rhodes et al, 2007, supra; Cancer Genome Atlas, supra), and accurately nominated the tumor suppressor IncRNA MEG3 (Rhodes et al, 2007, supra; Cancer Genome Atlas, supra ) and the metastasis suppressor LIFR (Chen, D. et al. Nature medicine 18, 1511-1517, (2012)) as highly negatively enriched (Fig. 3c-e). Similarly, in the prostate cancer set SSEA detected differential expression of IncRNAs and protein-coding genes consistent with the literature (Fig. 3f). Notably, the known prostate cancer IncRNAs Prostate Cancer Antigen-3 (PCA3) and SChLAPl were strikingly enriched in a cancer-specific and prostate-specific manner relative to all other sample set analyses (Fig. 3g,h) (Taylor et al, supra; Presner et al., 2013, supra). Overall the ability of the enrichment testing approach to rediscover known cancer genes in an unbiased fashion indicates its utility for the analysis of the cancer association and lineage specificity within the panorama of uncharacterized transcription unveiled by MiTranscriptome.
Characterization of lineage-specific and cancer -specific IncRNA transcription
To extend the study beyond known cancer genes, the enrichment test results for lineage- specific and cancer-specific transcripts were mined in an unbiased manner. Lineage specificity was assayed using sample sets for each cancer or tissue type compared to all other samples in the MiTranscriptome compendium (Figure 3a, "Cancer Types/Normal Types"), and SSEA results were utilized to determine the degree of enrichment for each transcript in the various cancer and tissue types. Unsupervised clustering of transcript percentile ranks for the top 1% of transcripts in each lineage demonstrated distinct signatures for each lineage while also described relationships among lineages and between cancer and normal sets from the same lineage (Fig. 14a). Examples of closely related lineage clusters include blood cancers (acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and myeloproliferative neoplasia (MPN)), brain cancers (lower grade glioma
(LGG) and glioblastome multiforme (GBM)), and muscle tissue (cardiac and skeletal). Additionally, a cluster comprising cervical cancer, head and neck cancer and normal lineages, lung squamous cell cancer, and bladder cancer emerged and indicated that primarily squamous (and transitional) cell carcinomas from distant primary sites share important gene expression relationships. Unsupervised clustering of only the IncRNAs in the top 1% of the SSEA analysis for lineage association recapitulated all of these relationships, indicating the capacity for IncRNAs to independently identify cancer and normal lineages (Fig. 4a).
Next, the dimension of cancer-specific transcriptional dynamics was investigated in twelve tissues with ample numbers of both cancer and normal samples (Figure 3a, "Cancer vs. Normal").
Similar to above, unsupervised clustering of the top 1% cancer-associated IncRNAs demonstrated highly specific signatures for each cancer type, with the exception of lung cancers and kidney cancers (Fig. 4b and Fig. 14b). Lung squamous cell carcinomas (LUSC) and adenocarcinomas (LUAD) clustered together and shared numerous transcripts with cancer association. Similarly, renal clear cell (KIRC) and papillary cell (KIRP) carcinomas exhibited highly overlapping signatures, while renal chromophobe carcinomas (KICH) remained distinct from KIRC and KIRP.
Finally, results from lineage and cancer analyses were intersected. Such transcripts have translational potential for use in non-invasive clinical tests, particularly for cancers that lack reliable biomarkers. Notable examples included the prostatespecific IncRNAs PCA3 and SChLAPl presented earlier (Fig. 3g,h). A myriad of IncRNAs were detected as being lineage and cancer associated (i.e. in the top 5% of both analyses) for each of the cancer types analyzed (Fig. 4c, Fig. 15a). A direct comparison of IncRNAs and protein-coding transcripts revealed that both annotated and unannotated IncRNAs have the perform at a comparable level to protein-coding genes in lineage and cancer association and support a role for IncRNAs as cancer specificity markers (Fig. 4d and Fig. 15b,c). After applying stringent statistical cutoffs to nominate the most compelling associations, a cohort of 7,942 IncRNA or TUCP genes (11,478 transcripts) were nominated as cancer associated, lineage associated, or both. Many of these IncRNAs also possessed base-wise conservation or ultraconserved elements (Fig. 2, Table 1). Transcripts meeting the stringent cutoffs in the cancer versus normal analyses ("Cancer vs. Normal", Fig. 3a) were designated as having "cancer association". Those transcripts meeting stringent cutoffs for linage specificity in non-cancerous tissue (e.g. heart, skeletal muscle, embryonic stem cells) and in cancers lacking RNA-Seq data for benign tissue were designated as "lineage associated". Moreover, transcripts meeting the cutoffs for both the cancer versus normal and lineage specificity analyses were designated as having "cancer and lineage association" (Table 1). Transcripts with significant association in just one tissue type were given names according to that tissue type (Table 1), and transcripts with associations in multiple tissues were named "Cancer Associated Transcripts" (CATs). An additional 545 IncRNA genes (1634 transcripts) that possessed ultraconserved elements but did not meet the stringent lineage and cancer association cutoffs were designated as HICLINCs (Highly Conserved Long Intergenic Non-Coding RNA). Taken together, the cancer and/or lineage IncRNAs and HICLINCs comprise a set of 8,487 IncRNAs that bear strong functional potential. 7,804 of these IncRNAs did not possess an official gene name according to the HUGO Gene Nomenclature Committee, and were thus given names according to the convention described above and in Table 1.
Additional analyses were performed to provide more information about these transcripts for use in selecting candidates for subsequent experimentation. A comprehensive assessment of
transcription factor binding to the promoters of these IncRNAs was performed using the ENCODE dataset for 161 transcription factors. Additionally, statistics describing the expression of each IncRNA in the different tissue cohorts is reported. For each TUCP transcript, the longest ORF, coding potential score, and presence of any pfam domain were identified.
Further interrogation of the relationship with GWAS SNPs was also performed, and all transcripts within 50kb of a GWAS SNP implicated in disease of the same cancer or tissue as the transcript awere identified. These IncRNAs provide candidates for intergenic expression quantitative trait loci (eQTLs) analysis. For example, the IncRNA named Breast Cancer Associated Transcript- 85, BRCAT49 is a breast cancer- and lineageassociated IncRNA (Fig. 4d) located ~45kb
downstream of a breast cancer SNP (rsl3387042) that has been implicated by six independent GWAS studies (Fig. 4f) (Li, J. et al. Breast cancer research and treatment 126, 717-727, (2011); Michailidou, K. et al. Nature genetics 45, 353-361, 361e351-352, (2013); Stacey, S. N. et al. Nature genetics 39, 865-869, (2007); Thomas, G. et al. Nature genetics 41, 579-584, doi: 10.1038/ng.353 (2009); Turnbull, C. et al. Nature genetics 42, 504-507, (2010)). The NHGRI GWAS catalog describes rsl3387042 as an intergenic SNP with no reported associated gene (Welter, D. et al.
Nucleic acids research 42, D1001-1006, (2014)). Given its breast cancer specificity (Fig. 4g), BRCAT49 provides a target for explaining the breast cancer association of this genomic region. Moreover, with further investigation and analysis, its cancer and lineage specificity support a role for BRCAT49 (and other similar cancer and lineage-specific IncRNAs) as a cancer specific
transcriptional marker. Additional representative expression profiles for cancer- or lineage-specific IncRNAs in other tissue types are displayed in Fig. 16c,d.
Because the MiTranscriptome represents such a comprehensive array of tissues and cancers (Fig. la), it is able to uncover an abundance of lineage and cancer specific transcription that has biological and clinical impact. A representative example of one such lineage specific IncRNA is a transcript was termed Melanoma Associated Transcript-7, MEAT6, which was found to be in the
99.8th percentile in the melanoma lineage SSEA analysis (Fig. 4a). Genomic investigation delineated MEAT6 as a partially annotated transcriptional variant of the UCSC IncRNA AK090788 IncRNA on chromosome 6q26 (Fig. 16a). However, MEAT6 utilizes an alternative start site and upstream exons absent from reference catalogs, highlighting the breadth and depth of transcriptome reconstruction effort. Expression of MEAT6 isoforms using the novel start site were highly specific to the melanoma samples in the MiTranscriptome cohort (Fig. 4e); however, isoforms lacking the MEAT6 start site had a dramatically different pan-cancer expression profile with almost no expression in melanoma (Fig. 16b). These findings manifest the ability of the assembly to provide a clear and consummate portrayal of the transcriptional activity that distinguishes disease types.
To further corroborate the differential expression analysis, a high-throughput "guilt- byassociation" analysis was performed for all of the IncRNAs meeting the stringent cutoffs.
Expression of each transcript isoform was correlated to all annotated protein-coding genes for each relevant tissue cohort, and various cancer signatures were tested for enrichment with the most correlated or anti-correlated genes using the GSEA method. The gene sets were curated and categorized into cancer relevant categories: angiogenesis/hypoxia associated, metastasis associated, proliferation/cell-cycle associated, adhesion associated, DNA damage/repair associate, oncogenic association, and miscellaneous cancer association. In total over 14 thousand transcripts were analyzed with this method, and the significantly associated cancer gene sets are reported (Tables 2 and 3).
Table 1. Summary of lineage and/or cancer- specific IncRNAs nominated in this study.

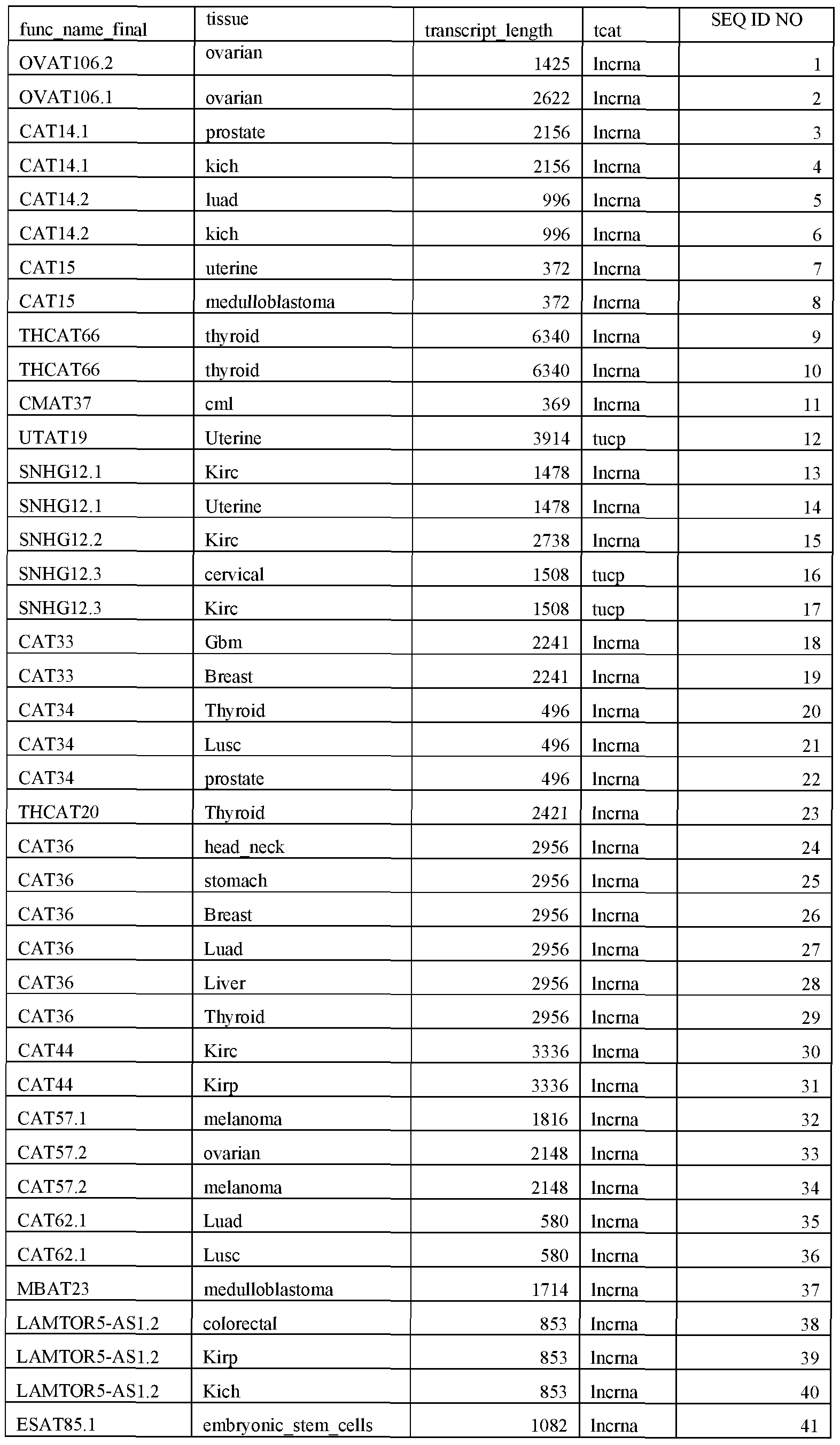
ESAT40 embryonic stem cells 787 tucp 43
THCAT22.1 Thyroid 2848 lncrna 44
THCAT22.1 Thyroid 2848 lncrna 45
THCAT22.4 Thyroid 2315 lncrna 46
THCAT22.4 Thyroid 2315 lncrna 47
THCAT22.3 Thyroid 3056 lncrna 48
THCAT22.3 Thyroid 3056 lncrna 49
CAT99.1 colorectal 3188 lncrna 50
CAT99.1 medulloblastoma 3188 lncrna 51
CAT99.2 Gbm 3526 lncrna 52
CAT99.2 Luad 3526 lncrna 5
CAT99.2 Lusc 3526 lncma 54
OVAT12 ovarian 653 lncrna 55
BRCAT23 Breast 2635 lncrna 56
CAT112.1 Luad 5210 lncrna 57
CAT112.2 cervical 6470 lncrna 58
CAT112.2 Luad 6470 lncrna 59
CAT112.2 skeletal muscle 6470 lncma 60
CAT115.1 head neck 1032 lncrna 61
CAT115.1 Lusc 1032 lncrna 62
CAT115.2 prostate 1599 lncrna 63
CAT118.1 Lgg 1658 tucp 64
RUSC1-AS1.1 Uterine 1852 tucp 65
RUSC1-AS1.1 Ami 1852 tucp 66
RUSC1-AS1.2 colorectal 7499 tucp 67
CAT122 head neck 9412 lncrna 68
CAT122 Kirp 9412 lncrna 69
CAT122 Liver 9412 lncrna 70
CAT122 Luad 9412 lncrna 71
CAT122 irc 9412 lncrna 72
PNAT1.2 pancreatic 902 lncrna 73
CAT147.1 medulloblastoma 1533 lncrna 74
MIR205HG.1 Lusc 4404 lncrna 75
MIR205HG.1 prostate 4404 lncrna 76
MIR205HG.2 Lusc 2753 tucp 77
MIR205HG.2 prostate 2753 tucp 78
MIR205HG.3 Lusc 2336 lncrna 79
MIR205HG.3 prostate 2336 lncma 80
CAT171.1 Breast 741 lncrna 81
CAT171.1 Luad 741 lncma 82
CAT179 Thyroid 475 lncrna 83
CAT179 Lusc 475 lncrna 84
CAT179 Kirp 475 lncrna 85
CAT179 Breast 475 lncrna 86
CAT179 Luad 475 lncrna 87
CAT179 prostate 475 lncrna 88
CAT179 Kich 475 lncrna 89
CAT186.1 Luad 3863 lncrna 90
CAT186.2 prostate 1661 lncrna 91
CAT186.2 Breast 1661 lncrna 92
CAT186.2 Luad 1661 lncrna 93
CAT186.2 Lusc 1661 lncrna 94
CAT186.2 1661 lncrna 95
CAT187.1 Heart 4840 lncrna 96
CAT187.2 Ami 9637 lncrna 97
CAT187.2 Kirc 9637 lncrna 98
ESAT33.2 embryonic stem cells 13412 lncrna 99
ESAT33.1 embryonic stem cells 2024 lncrna 100
CAT1224.1 head neck 2502 lncrna 101
CALML3-AS1.1 prostate 11669 lncrna 102
CALML3-AS1.1 Breast 11669 lncrna 103
CALML3-AS1.2 Lusc 8709 tucp 104
GATA3-AS1.1 Kirc 7280 lncrna 105
GATA3-AS1.2 Breast 9735 lncrna 106
BRCAT1.1 Breast 6529 lncrna 107
BRCAT1.1 Breast 6529 lncrna 108
BRCAT1.4 Breast 26917 lncrna 109
BRCAT1.4 Breast 26917 lncrna 110
BRCAT1.3 Breast 1676 lncrna 111
BRCAT1.3 Breast 1676 lncrna 112
BRCAT1.5 Breast 8975 lncrna 113
BRCAT1.5 Breast 8975 lncrna 114
BRCAT1.2 Breast 32732 lncrna 115
BRCAT1.2 Breast 32732 lncrna 116
CAT1233 Thyroid 1709 lncrna 117
CAT1233 pancreatic 1709 lncrna 118
CAT1235.1 melanoma 3656 lncrna 119
CAT1235.1 Kirc 3656 lncrna 120
CAT1235.1 Kirp 3656 lncrna 121
CAT1235.1 ovarian 3656 lncrna 122
CAT1235.1 prostate 3656 lncrna 123
CAT1235.1 Breast 3656 lncrna 124
CAT1235.1 Lusc 3656 lncrna 125
CAT1235.2 Uterine 4480 lncrna 126
CAT1235.2 Kirc 4480 lncrna 127
CAT1235.2 cerv ical 4480 lncrna 128
CAT1235.2 Breast 4480 lncrna 129
CAT1235.2 Luad 4480 lncrna 1 0
CAT1235.2 Lusc 4480 lncrna 131
ST8SIA6-AS1.1 prostate 1250 lncrna 132
ST8SIA6-AS1.1 Liver 1250 lncrna 133
ST8SIA6-AS1.2 prostate 10110 lncrna 134
ST8SIA6-AS1.2 Liver 10110 lncrna 135
LINC00948.1 pancreatic 1205 lncrna 136
LINC00948.1 Kirp 1205 lncrna 137
CAT1269 Mpn 9975 lncrna 138
CAT1269 Cml 9975 lncrna 139
CAT1269 medulloblastoma 9975 lncrna 140
UNC5B-AS1.1 ovarian 1602 lncrna 141
UNC5B-AS1.1 Thyroid 1602 lncrna 142
UNC5B-AS1.2 Thyroid 915 lncrna 143
UNC5B-AS1.2 prostate 915 lncrna 144
KCCAT243 Kirc 1647 lncrna 145
KCCAT243 Kirc 1647 lncrna 146
OVAT44 ovarian 1283 lncrna 147
CAT1284.1 Kirc 17882 lncrna 148
CAT1284.1 Kirp 17882 lncrna 149
CAT1284.1 Lusc 17882 lncrna 150
CAT1284.1 pancreatic 17882 lncrna 151
CAT1284.1 prostate 17882 lncrna 152
CAT1284.1 Kich 17882 lncrna 153
MEAT20.3 melanoma 1252 lncrna 154
MEAT20.1 melanoma 1222 lncrna 155
MEAT20.2 melanoma 1028 lncrna 156
CAT 1324 colorectal 1329 tucp 157
CAT 1324 Uterine 1329 tucp 158
CAT 1324 head neck 1329 tucp 159
CMAT6 Cml 540 lncrna 160
CAT1337.1 Lusc 41069 lncrna 161
CAT1337.1 pancreatic 41069 lncrna 162
CAT1337.1 prostate 41069 lncrna 163
LINC00958.1 Lusc 42272 lncrna 164
LINC00958.1 prostate 42272 lncrna 165
LINC00958.2 Lusc 38359 lncrna 166
CAT1337.2 head neck 2101 lncrna 167
CAT1337.2 Lusc 2101 lncrna 168
CAT1337.2 pancreatic 2101 lncrna 169
CAT1337.2 medulloblastoma 2101 lncrna 170
CAT1337.2 prostate 2101 lncrna 171
LINC00958.3 Lusc 30635 lncrna 172
LINC00958.3 pancreatic 30635 lncrna 173
LINC00958.4 Lusc 14161 lncrna 174
LINC00958.4 pancreatic 14161 lncrna 175
LINC00958.5 Luad 8665 lncrna 176
CAT1345.1 Luad 1080 lncrna 177
CAT1345.1 Lusc 1080 lncrna 178
LINC00678.1 embn onic stem cells 471 lncrna 179
WT1-AS.1 ovarian 4079 lncrna 180
WT1-AS.2 ovarian 132 lncrna 181
WT1-AS.3 ovarian 3618 lncrna 182
WT1-AS.3 Kich 3618 lncrna 183
CAT1363.1 Uterine 2421 lncrna 184
CAT1363.2 Uterine 2263 lncrna 185
CAT1363.2 Kich 2263 lncrna 186
CAT1363.2 Lusc 2263 lncrna 187
UTAT10 Uterine 1287 tucp 188
NEAT1 Kich 26332 lncrna 189
MALAT1.2 Heart 7744 lncrna 190
MALAT1.2 Kich 7744 lncrna 191
CAT1373 Breast 3975 lncrna 192
CAT 1373 Luad 3975 lncrna 193
CAT 1373 Lusc 3975 lncrna 194
CAT 1373 skeletal muscle 3975 lncrna 195
CAT 1373 Gbm 3975 lncrna 196
CAT 1373 Lgg 3975 lncrna 197
CAT1373 meduUoblastoma 3975 lncrna 198
CAT1373 Kirc 3975 lncrna 199
CAT 1376 Uterine 8904 tucp 200
CAT 1376 Kich 8904 tucp 201
CAT 1376 meduUoblastoma 8904 tucp 202
OVAT47 ovarian 833 lncrna 203
ANOl-ASl head neck 10628 tucp 204
ANOl-ASl Kirc 10628 tucp 205
ANOl-ASl Cml 10628 tucp 206
ANOl-ASl Gbm 10628 tucp 207
ANOl-ASl Lgg 10628 tucp 208
ANOl-ASl meduUoblastoma 10628 tucp 209
ANOl-ASl prostate 10628 tucp 210
CAT1385.1 Thyroid 8545 lncrna 211
CAT1385.1 head neck 8545 lncrna 212
CAT1385.2 Thyroid 3095 lncrna 213
CAT1385.2 head neck 3095 lncrna 214
CAT1385.2 Breast 3095 lncrna 215
CAT1385.2 Luad 3095 lncrna 216
CAT1385.2 Ami 3095 lncrna 217
CAT1385.3 medulloblastoma 4221 lncrna 218
CAT13 1 Cml 7090 lncrna 219
CAT1391 Ami 7090 lncrna 220
CAT1391 Kirc 7090 lncrna 221
CAT1399 Thyroid 6255 lncrna 222
CAT 1399 stomach 6255 lncrna 223
CAT 1399 head neck 6255 lncrna 224
CAT 1399 Kirp 6255 lncrna 225
CAT 1399 Luad 6255 lncrna 226
CAT 1399 Kirc 6255 lncrna 227
CAT 1399 Mpn 6255 lncrna 228
CAT1399 Ami 6255 lncrna 229
CAT 1399 Lgg 6255 lncrna 230
CAT1425.1 Kirc 1151 lncrna 231
CAT1425.1 Lusc 1151 lncrna 232
CAT1425.1 Breast 1151 lncrna 233
CAT1425.1 Liver 1151 lncrna 234
CAT1425.2 Kirc 5627 lncrna 235
CAT1425.2 Breast 5627 lncrna 236
CAT1425.2 Liver 5627 lncrna 237
CAT1434 ovarian 637 lncrna 238
CAT1434 Uterine 637 lncma 239
CAT1434 Kich 637 lncrna 240
ESAT80 embryonic stem cells 4806 lncrna 241
CAT1452.1 Kich 3332 tucp 242
CAT1452.1 prostate 3332 tucp 243
CAT1452.1 Luad 3332 tucp 244
CAT1452.1 Lusc 3332 tucp 245
CAT 1464 Kich 7157 lncrna 246
CAT 1464 Gbm 7157 lncrna 247
CAT 1464 melanoma 7157 lncrna 248
CAT 1464 Lgg 7157 lncrna 249
ESAT3 embryonic stem cells 2056 lncma 250
ME AT 11 melanoma 1006 lncrna 251
CAT1468 Kirc 2243 lncrna 252
CAT 1468 medulloblastoma 2243 lncrna 253
CAT 1469 head neck 2125 lncrna 254
CAT 1469 Lusc 2125 lncrna 255
CAT1472.1 Lusc 1709 lncrna 256
DDX11-AS1.1 Kirp 2011 lncrna 257
DDX11-AS1.1 Liver 2011 lncrna 258
DDX11-AS1.1 Lusc 2011 lncrna 259
CAT1501.1 Kich 3920 lncrna 260
CAT1501.1 Thyroid 3920 lncrna 261
CAT1501.2 ovarian 4652 lncrna 262
CAT1528 Heart 2528 lncrna 263
CAT1528 Kich 2528 lncrna 264
CAT1528 Thyroid 2528 lncrna 265
MEAT 10 melanoma 1589 lncrna 266
CRAT16 colorectal 1650 lncrna 267
HNCAT4 head neck 764 lncrna 268
CAT1547 Mpn 3302 lncrna 269
CAT1547 Cml 3302 lncrna 270
CAT1547 Kirc 3302 lncrna 271
CAT1547 embryonic stem cells 3302 lncrna 272
CAT1547 skeletal muscle 3302 lncrna 273
CAT1564 Ami 4645 tucp 274
CAT1564 Kirc 4645 tucp 275
ESAT56.3 embryonic stem cells 8304 lncrna 276
ESAT56.2 embryonic stem cells 7622 lncrna 277
ESAT56.1 embryonic stem cells 7755 lncrna 278
LINC00428 embryonic stem cells 7715 lncrna 279
LINC00458.2 embryonic stem cells 2657 lncrna 280
LINC00458.1 embryonic_stem_cells 1021 lncrna 281
ESAT23 embryonic stem cells 1747 lncrna 282
LINC00458.3 embryonic stem cells 2948 lncrna 283
ESAT13.3 embryonic stem cells 1855 lncrna 284
ESAT13.1 embryonic stem cells 878 lncrna 285
ESAT34 embryonic stem cells 923 lncrna 286
ESAT36.1 embryonic stem cells 10586 lncrna 287
ESAT36.4 embryonic stem cells 11133 lncrna 288
ESAT36.2 embryonic stem cells 11103 lncrna 289
ESAT36.3 embryonic stem cells 13928 lncrna 290
MEAT69 melanoma 2031 lncrna 291
DOCK9-AS2 thyroid 2091 lncrna 292
DOCK9-AS2 thyroid 2091 lncrna 293
CAT1629.1 medulloblastoma 5852 lncrna 294
CAT1629.2 hiad 3834 lncrna 295
CAT1629.2 lusc 3834 lncrna 296
CAT1629.2 pancreatic 3834 lncrna 297
CAT1629.2 kirc 3834 lncrna 298
CAT1631 kirc 1210 lncrna 299
CAT1631 medulloblastoma 1210 lncrna 300
GBAT25.1 gbm 3520 lncrna 301
SMAT25 skeletal muscle 6343 tucp 302
CAT1636.1 head neck 1465 lncrna 303
CAT1636.1 uterine 1465 lncrna 304
CAT1641 colorectal 3474 lncrna 305
CAT1641 stomach 3474 lncrna 306
CAT1641 kich 3474 lncrna 307
CAT1641 liver 3474 lncrna 308
UTAT29 uterine 322 lncrna 309
OVAT48 ovarian 650 lncrna 310
ESAT39.3 embryonic stem cells 8927 lncrna 311
CAT1658 head neck 835 lncrna 312
CAT1658 lusc 835 lncrna 313
CAT1659.1 breast 1250 lncrna 314
CAT1659.2 embryonic stem cells 15089 lncrna 315
CAT1659.2 breast 15089 lncrna 316
CAT1659.3 embryonic stem cells 1255 lncrna 317
CAT1659.4 embryonic stem cells 13866 lncrna 318
CAT1683.1 kich 1006 lncrna 319
CAT1683.1 embryonic stem cells 1006 lncrna 320
OVAT5 ovarian 288 lncrna 321
CAT1723 ami 2435 tucp 322
CAT1723 head neck 2435 tucp 323
CAT1723 luad 2435 tucp 324
CAT1723 lusc 2435 tucp 325
CAT1723 gbm 2435 tucp 326
CAT1728 heart 3209 tucp 327
CAT1728 thyroid 3209 tucp 328
CAT1728 breast 3209 tucp 329
CAT1728 luad 3209 tucp 330
CAT1728 lusc 3209 tucp 331
CAT1735 breast 850 lncrna 332
CAT1735 luad 850 lncrna 333
CAT1735 lusc 850 lncrna 334
CAT1735 thyroid 850 lncrna 335
CAT1735 kirp 850 lncrna 336
CAT1736.1 thyroid 1229 lncrna 337
CAT1736.1 luad 1229 lncrna 338
CAT1760.1 melanoma 6162 lncrna 339
CAT1760.1 igg 6162 lncrna 340
CAT1760.1 prostate 6162 lncrna 341
CAT1760.2 kirc 7244 lncrna 342
CAT1760.2 melanoma 7244 lncrna 343
USP3 -AS1.3 colorectal 477 lncrna 344
USP3 -AS1.3 uterine 477 lncrna 345
USP3-AS1.3 melanoma 477 lncrna 346
CAT 1768.1 prostate 6771 lncrna 347
CAT1768.1 kirp 6771 lncrna 348
CAT1773 melanoma 834 lncrna 349
CAT1773 thyroid 834 lncrna 350
CAT1773 kirc 834 lncrna 351
CAT1773 liver 834 lncrna 352
CAT1773 cml 834 lncrna 353
CAT1777 thyroid 1 1399 tucp 354
CAT1777 cervical 1 1399 tucp 355
CAT1777 mad 1 1399 tucp 356
CAT1777 hisc 1 1399 tucp 357
CAT1784 IRS 2795 lncrna 358
CAT1784 head neck 2795 lncrna 359
CAT1784 kirp 2795 lncrna 360
CAT1784 breast 2795 lncrna 361
CAT1784 kirc 2795 lncrna 362
UTAT4 uterine 650 lncrna 363
CAT1785 heart 852 lncrna 364
CAT1785 embryonic stem cells 852 lncrna 365
CAT1796 thyroid 3587 lncrna 366
CAT1796 prostate 3587 lncrna 367
CAT1796 breast 3587 lncrna 368
KCCAT209 kirc 3196 lncrna 369
KCCAT1 1.2 kirc 2905 lncrna 370 CCAT1 1.2 kirc 2905 lncrna 371
KCCAT1 1.1 kirc 1577 lncrna 372
KCCAT1 1.1 kirc 1577 lncrna 373
AMAT59 ami 4948 lncrna 374
KCCAT71 kirc 1442 lncrna 375
CAT 1825 thyroid 1672 lncrna 376
CAT 1825 kich 1672 lncrna 377
CAT 1825 igg 1672 lncrna 378
CAT1825 embryonic stem cells 1672 lncrna 379
CAT 1837.1 cervical 605 lncrna 380
CAT1837.2 ami 1853 lncrna 381
CAT1841.1 kirc 2864 lncrna 382
CAT1841.1 kirp 2864 lncrna 383
CAT1841.1 breast 2864 lncrna 384
CAT1841.1 ovarian 2864 lncrna 385
CAT1841.1 skeletal muscle 2864 lncrna 386
CAT1841.1 uterine 2864 lncrna 387
CAT 1843 kirc 1519 lncrna 388
CAT1843 liver 1519 lncrna 389
CAT1844 igg 2653 lncrna 390
CAT 1844 thyroid 2653 lncrna 391
CRNDE.l kirp 10743 lncrna 392
CRNDE.2 colorectal 10057 lncrna 393
CAT1871.1 gbm 5165 lncrna 394
CAT1871.1 5165 lncrna 395
CAT1871.2 7095 lncrna 396
VPS9D1-AS1.1 prostate 2428 lncrna 397
VPS9D1-AS1.1 lusc 2428 lncrna 398
VPS9D1-AS1.2 prostate 3100 lncrna 399
VPS9D1-AS1.2 luad 3100 lncrna 400
VPS9D1-AS1.2 lusc 3100 lncrna 401
VPS9D1-AS1.3 prostate 2710 lncrna 402
VPS9D1-AS1.3 lusc 2710 lncrna 403
CAT1889 melanoma 925 lncrna 404
CAT1889 head neck 925 lncrna 405
CAT1889 kirp 925 lncrna 406
CAT1889 breast 925 lncrna 407
CAT1889 luad 925 lncrna 408
PITPNA-AS1 gbm 356 lncrna 409
CAT1892.1 uterine 2087 lncrna 410
CAT1893 ovarian 990 lncrna 411
CAT1893 kich 990 lncrna 412
CAT1909.1 gbm 2569 tucp 413
CAT 1909.1 colorectal 2569 tucp 414
CAT1909.1 uterine 2569 tucp 415
CAT 1909.1 liver 2569 tucp 416
CAT1909.2 pancreatic 3180 tucp 417
CAT1909.2 cervical 3180 tucp 418
CAT1909.2 kirc 3180 tucp 419
CAT1915 uterine 1314 lncrna 420
CAT1915 cml 1314 lncrna 421
CAT1928 colorectal 4160 tucp 422
CAT1928 thyroid 4160 tucp 423
CAT 1928 head neck 4160 tucp 424
CAT 1928 kirp 4160 tucp 425
CAT 1940 gbm 578 lncrna 426
CAT 1940 kirp 578 lncrna 427
CAT1940 kich 578 lncrna 428
CAT 1944 uterine 749 lncrna 429
CAT 1944 luad 749 lncrna 430
CAT 1949 colorectal 2817 lncrna 431
CAT1949 uterine 2817 lncrna 432
CAT 1949 rapn 2817 lncrna 433
CAT 1949 kirc 2817 lncrna 434
MPAT8 rapn 952 lncrna 435
CAT1957.1 lusc 1013 lncrna 436
CAT1957.1 head neck 1013 lncrna 437
CAT1957.1 breast 1013 lncrna 438
CAT1957.1 luad 1013 lncrna 439
CAT1957.1 kirc 1013 lncrna 440
CAT1957.1 liver 1013 lncrna 441
CAT 1964.1 kirc 9319 lncrna 442
CAT 1964.1 cervical 9319 lncrna 443
CAT 1964.1 medulloblastoma 9319 lncrna 444
CAT 1964.1 breast 9319 lncrna 445
CAT 1964.1 luad 9319 lncrna 446
CAT 1964.1 lusc 9319 lncrna 447
CAT1964.2 kirc 3949 lncrna 448
CAT1964.2 embryonic stem cells 3949 lncrna 449
CAT1964.2 ovarian 3949 lncrna 450
CAT1964.2 medulloblastoma 3949 lncrna 451
CAT1964.2 breast 3949 lncrna 452
CAT1964.2 luad 3949 lncrna 453
CAT1964.2 lusc 3949 lncrna 454
CAT 1967.1 lusc 2747 lncrna 455
CAT 1967.1 mpn 2747 lncrna 456
CAT1967.1 thyroid 2747 lncrna 457
CAT 1967.1 prostate 2747 lncrna 458
CAT 1968.1 kirc 17122 tucp 459
CAT1968.2 gbra 3663 tucp 460
CAT1968.2 igs 3663 tucp 461
CAT1968.2 colorectal 3663 tucp 462
CAT1968.2 prostate 3663 tucp 463
LINC00511.1 luad 13352 tucp 464
LINC00511.2 lusc 7531 lncrna 465
LINC00511.3 thyroid 5973 lncrna 466
LINC00511.3 luad 5973 lncrna 467
LINC00511.3 lusc 5973 lncrna 468
CAT 1977 gbra 1001 lncrna 469
CAT 1977 melanoma 1001 lncrna 470
CAT 1977 igg 1001 lncrna 471
CAT 1977 kirc 1001 lncrna 472
MEAT77 melanoma 1767 lncrna 473
MEAT75 melanoma 4785 tucp 474
CAT1984 uterine 3024 lncrna 475
CAT1984 thyroid 3024 lncrna 476
CAT1984 breast 3024 lncrna 477
CAT1984 liver 3024 lncrna 478
UTAT3 uterine 2710 tucp 479
MAFG-AS1.1 lusc 4227 tucp 480
MAFG-AS1.1 kirp 4227 tucp 481
MAFG-AS1.1 breast 4227 tucp 482
MAFG-AS1.1 luad 4227 tucp 483
MAFG-AS1.1 prostate 4227 tucp 484
MAFG-AS1.1 liver 4227 tucp 485
MAFG-AS1.1 mpn 4227 tucp 486
MAFG-AS1.2 breast 4434 tucp 487
MAFG-AS1.2 luad 4434 tucp 488
MAFG-AS1.2 lusc 4434 tucp 489
LINC00668.1 head neck 3547 lncrna 490
LINC00668.1 lusc 3547 lncrna 491
LINC00668.2 lusc 3153 lncrna 492
UTAT6 uterine 532 lncrna 493
LINC00669 luad 439 lncrna 494
LINC00669 lusc 439 lncrna 495
ESAT59 embryonic stem cells 954 lncrna 496
CAT2045.1 lusc 4504 lncrna 497
CAT2045.2 colorectal 825 lncrna 498
CAT2045.2 kich 825 lncrna 499
CAT2045.2 cml 825 lncrna 500
CAT2045.2 ami 825 lncrna 501
CAT2045.2 kirp 825 lncrna 502
CAT2045.2 luad 825 lncrna 503
MIR7-3HG.7 pancreatic 3602 lncrna 504
MIR7-3HG.10 pancreatic 3029 lncrna 505
MIR7-3HG.22 pancreatic 3219 lncrna 506
MIR7-3HG.6 pancreatic 2963 lncrna 507
MIR7-3HG.19 pancreatic 2698 lncrna 508
MIR7-3HG.16 pancreatic 3067 lncrna 509
MIR7-3HG.20 pancreatic 2737 lncrna 510
MIR7-3HG.23 pancreatic 3021 lncrna 511
MIR7-3HG.3 pancreatic 2895 lncrna 512
MIR7-3HG.1 pancreatic 3596 lncrna 513
MIR7-3HG.2 pancreatic 3598 lncrna 514
MIR7-3HG.18 pancreatic 3581 lncrna 515
CAT2059 kirc 8226 lncrna 516
CAT2059 kirc 8226 lncrna 517
CAT2059 breast 8226 lncrna 518
CAT2069.1 colorectal 786 tucp 519
UTAT37 uterine 1498 lncrna 520
CAT2082.1 embryonic stem cells 8633 tucp 521
CAT2082.1 breast 8633 tucp 522
CAT2082.1 cervical 8633 tucp 523
LINC00906.1 kich 27059 lncrna 524
CAT2092.1 kich 3077 lncrna 525
CAT2092.1 thyroid 3077 lncrna 526
CAT2092.2 kich 2061 lncrna 527
CAT2092.2 thyroid 2061 lncrna 528
LINC00906.2 kich 36082 tucp 529
CAT2095 kirc 11872 tucp 530
CAT2095 kich 11872 tucp 531
MBAT15 medulloblastoma 5173 lncrna 532
LINC00665.1 prostate 1615 lncrna 533
LINC00665.1 luad 1615 lncrna 534
LINC00665.1 colorectal 1615 lncrna 535
LINC00665.2 prostate 6207 lncrna 536
LINC00665.2 colorectal 6207 lncrna 537
LINC00665.3 embryonic stem cells 1519 lncrna 538
LINC00665.4 prostate 4249 lncrna 539
LINC00665.5 prostate 10952 lncrna 540
LINC00665.5 colorectal 10952 lncrna 541
HRAT8 heart 333 lncrna 542
CAT2118.1 thyroid 1437 lncrna 543
CAT2118.1 breast 1437 lncrna 544
CAT2118.1 breast 1437 lncrna 545
CAT2118.1 ovarian 1437 lncrna 546
CAT2118.1 medulloblastoma 1437 lncrna 547
CAT2118.2 thyroid 2401 lncrna 548
CAT2118.2 breast 2401 lncrna 549
CAT2120 cml 2146 lncrna 550
CAT2120 kirc 2146 lncrna 551
CAT2120 embryonic stem cells 2146 lncrna 552
CAT2120 luad 2146 lncrna 553
CAT2120 lusc 2146 lncrna 554
HNCAT3.1 head neck 6262 lncrna 555
CAT201.1 embryonic_stem_cells 3565 lncrna 556
CAT201.2 kich 1898 lncrna 557
MEAT48.1 melanoma 2063 lncrna 558
CAT219.1 colorectal 4360 lncrna 559
CAT219.1 uterine 4360 lncrna 560
CAT219.2 head neck 4284 lncrna 561
CAT219.2 lusc 4284 lncrna 562
CAT219.3 colorectal 7435 lncrna 563
CAT219.3 uterine 7435 lncrna 564
CAT226.1 melanoma 1445 lncrna 565
CAT226.2 melanoma 2173 lncrna 566
CAT226.3 melanoma 2427 lncrna 567
CAT226.4 kich 12408 lncrna 568
CAT226.5 kirc 7403 lncrna 569
CAT226.5 kirc 7403 lncrna 570
CAT226.6 kirc 27140 lncrna 571
CAT226.6 kirc 27140 lncrna 572
CAT227.1 kirc 2216 lncrna 573
CAT227.1 kirc 2216 lncrna 574
CAT227.2 kirc 8010 lncrna 575
CAT227.2 kirc 8010 lncrna 576
CAT227.3 kirc 21694 lncrna 577
CAT227.3 kirc 21694 lncrna 578
CAT227.3 kich 21694 lncrna 579
OVAT 114.6 ovarian 1428 lncrna 580
CAT249.1 skeletal muscle 1566 lncrna 581
CAT249.1 kich 1566 lncrna 582
CAT249.1 breast 1566 lncrna 583
CAT252.1 liver 660 lncrna 584
CAT255.1 head neck 2682 tucp 585
CAT255.1 luad 2682 tucp 586
CAT255.1 lusc 2682 tucp 587
CAT255.1 gbm 2682 tucp 588
CAT255.2 lusc 10836 lncrna 589
LINC00152.1 head neck 17432 lncrna 590
LINC00152.1 stomach 17432 lncrna 591
LINC00152.2 luad 3471 lncrna 592
GBAT19 gbm 736 lncrna 593
MIR4435-1HG.1 stomach 7049 lncrna 594
MIR4435-1HG.2 kirc 7670 lncrna 595
CAT313.1 kirc 4601 lncrna 596
CAT313.2 colorectal 414 lncrna 597
CAT313.3 kirc 1782 lncrna 598
CAT313.4 kirc 3674 lncrna 599
CAT313.5 kirc 3554 lncrna 600
CAT313.5 lusc 3554 lncrna 601
CERS6-AS1 breast 3097 lncrna 602
CERS6-AS1 luad 3097 lncrna 603
CERS6-AS1 lusc 3097 lncrna 604
CERS6-AS1 breast 3097 lncrna 605
PRCAT44 prostate 1046 lncrna 606
PRCAT44 prostate 1046 lncrna 607
HOXD-AS1 liver 3808 lncrna 608
HOXD-AS1 kirc 3808 lncrna 609
HOXD-AS1 kirp 3808 lncrna 610
HOXD-AS1 lusc 3808 lncrna 611
TTN-AS1.2 heart 1538 lncrna 612
TTN-AS1.3 skeletal muscle 1559 lncma 613
PRCAT122 prostate 4816 lncrna 614
PRCAT122 prostate 4816 lncrna 615
ESAT86 embryonic stem cells 8125 lncrna 616
CAT350 kirc 2633 lncrna 617
CAT350 medulloblastoma 2633 lncrna 618
CAT350 luad 2633 lncrna 619
CAT355.1 colorectal 5090 lncrna 620
CAT355.1 uterine 5090 lncrna 621
CAT355.1 head neck 5090 lncrna 622
CAT355.2 heart 4143 lncrna 623
CAT355.2 pancreatic 4143 lncrna 624
CAT355.2 head neck 4143 lncrna 625
CAT359.1 kich 774 lncrna 626
CAT359.1 kich 774 lncrna 627
CAT359.1 kirp 774 lncrna 628
CAT366.1 thyroid 1533 lncrna 629
PNAT34 pancreatic 1634 lncrna 630
ESAT94.1 embryonic stem cells 7212 lncrna 631
OVAT30 ovarian 2756 lncrna 632
CAT2158 embryonic stem cells 5319 lncrna 633
CAT2158 head neck 5319 lncrna 634
CAT2158 stomach 5319 lncrna 635
CAT2160.1 head neck 478 lncrna 636
CAT2160.2 kich 511 lncrna 637
CAT2160.2 pancreatic 511 lncrna 638
CAT2160.2 medulloblastoma 511 lncrna 639
CAT2160.2 lusc 511 lncrna 640
PNAT11.3 pancreatic 10140 lncrna 641
PNAT11.2 pancreatic 7906 lncrna 642
PNAT11.1 pancreatic 8471 lncrna 643
PNAT11.4 pancreatic 8912 lncrna 644
OSER1-AS1.1 gbm 2198 lncrna 645
OSER1-AS1.1 2198 lncrna 646
OSER1-AS1.2 uterine 4658 lncrna 647
OSER1-AS1.3 mpn 4342 lncrna 648
CAT2176.1 head neck 1893 lncrna 649
ZFAS1.1 thyroid 2921 lncrna 650
ZFAS1.1 kirc 2921 lncrna 651
ZFAS1.2 kirc 3006 lncrna 652
ZFAS1.2 kirp 3006 lncrna 653
ZFAS1.3 thyroid 4202 lncrna 654
ZFAS1.3 kirc 4202 lncrna 655
ZFAS1.3 heart 4202 lncrna 656
ZFAS1.3 uterine 4202 lncrna 657
ZFAS1.4 erabn onic stem cells 5507 lncrna 658
ZFAS1.4 kirc 5507 lncrna 659
CAT2180.1 luad 1833 lncrna 660
CAT2180.2 luad 1960 tucp 661
CAT2180.2 lusc 1960 tucp 662
MEAT44 melanoma 1645 lncrna 663
CAT2186.1 melanoma 1000 lncrna 664
OVAT65.1 ovarian 1284 lncrna 665
OVAT65.2 ovarian 1052 lncrna 666
OVAT65.4 ovarian 867 lncrna 667
IL10RB-AS1.2 kirc 3951 lncrna 668
PRCAT38 prostate 2598 lncrna 669
PRCAT38 prostate 2598 lncrna 670
PRCAT23 prostate 98 lncrna 671
PRCAT23 prostate 98 lncrna 672
CAT2215.1 melanoma 10845 lncrna 673
CAT2215.2 prostate 9282 lncrna 674
UTAT36 uterine 732 lncrna 675
DGCR5.1 kirc 12734 tucp 676
DGCR10.1 kirc 19778 tucp 677
DGCR10.1 kirc 19778 tucp 678
DGCR5.2 kirc 6204 tucp 679
DGCR5.2 kirp 6204 tucp 680
DGCR5.2 kirc 6204 tucp 681
DGCR5.3 kirc 1170 lncrna 682
DGCR5.3 kirc 1170 lncrna 683
DGCR10.2 kirc 18135 tucp 684
DGCR5.4 kirc 2794 tucp 685
DGCR5.4 kirc 2794 tucp 686
DGCR5.5 kirc 13853 tucp 687
DGCR5.5 kirc 13853 tucp 688
DGCR5.6 kirc 1384 lncrna 689
DGCR5.6 kirc 1384 lncrna 690
PRCAT104.4 prostate 6446 tucp 691
PRCAT104.4 prostate 6446 tucp 692
PRCAT104.3 prostate 11062 tucp 693
PRCAT104.3 prostate 11062 tucp 694
PRCAT104.6 prostate 2283 lncrna 695
PRCAT104.6 prostate 2283 lncrna 696
TUG1.1 thyroid 8722 lncrna 697
TUG1.2 thyroid 8736 lncrna 698
TUG 1.3 head neck 8597 lncrna 699
KHCAT21.2 kich 4229 lncrna 700
KHCAT21.1 kich 4310 lncrna 701
ESAT27 embryonic stem cells 908 lncrna 702
CAT2248 kirc 1515 lncrna 703
CAT2248 kirp 1515 lncrna 704
CAT2248 prostate 1515 lncrna 705
CAT2248 breast 1515 lncrna 706
CAT2251.1 luad 6597 lncrna 707
HRAT18 heart 546 lncrna 708
FGD5-AS1.2 gbra 3826 tucp 709
FGD5-AS1.2 igg 3826 tucp 710
FGD5-AS1.2 kirc 3826 tucp 711
FGD5-AS1.2 prostate 3826 tucp 712
FGD5-AS1.2 luad 3826 tucp 713
FGD5-AS1.2 lusc 3826 tucp 714
FGD5-AS1.3 kirc 3840 tucp 715
FGD5-AS1.4 luad 3882 tucp 716
LVCAT12 liver 4522 lncrna 717
GBAT1 gbm 1849 lncrna 718
ESRG.l embryonic stem cells 4629 lncrna 719
ESRG.l lusc 4629 lncrna 720
ESRG.2 embryonic stem cells 7300 lncrna 721
ESRG.3 ovarian 5685 lncrna 722
ESRG.4 ovarian 10753 lncrna 723
ESRG.5 ovarian 6373 lncrna 724
H1FX-AS1.1 ovarian 3535 lncrna 725
H1FX-AS1.1 uterine 3535 lncrna 726
ESAT16.4 embryonic stem cells 3076 lncrna 727
CAT474.1 embryonic stem cells 5036 tucp 728
ESAT19.2 embryonic stem cells 1639 lncrna 729
ESAT19.3 embryonic stem cells 870 lncrna 730
ESAT19.1 embryonic_stem_cells 996 lncrna 731
MFI2-AS1.1 lusc 1268 lncrna 732
MFI2-AS1.2 lusc 1504 lncrna 733 PCAT2.1 kirp 6514 tucp 734
LINC00504 ami 673 lncrna 735
LINC00504 head neck 673 lncrna 736
LINC00504 luad 673 lncrna 737
LINC00504 lusc 673 lncrna 738
KCCAT215 kirc 6566 lncrna 739
UGDH-AS1.3 ami 5875 tucp 740
UGDH-AS1.3 lusc 5875 tucp 741
CAT558.1 luad 837 lncrna 742
CAT558.1 lusc 837 lncrna 743
CAT558.1 prostate 837 lncrna 744
CAT558.2 lusc 757 lncrna 745
CAT565 thyroid 1574 lncrna 746
CAT565 luad 1574 lncrna 747
CAT565 cml 1574 lncrna 748
CAT565 gbm 1574 lncrna 749
CAT565 melanoma 1574 lncrna 750
CAT565 skeletal muscle 1574 lncrna 751
CAT565 medulloblastoma 1574 lncrna 752
CAT565 liver 1574 lncrna 753
LINC01094 gbm 10936 lncrna 754
LINC01094 10936 lncrna 755
LINC01094 kirc 10936 lncrna 756
LINC01094 kirp 10936 lncrna 757
LINC01094 breast 10936 lncrna 758
CAT566.1 gbm 11030 lncrna 759
CAT566.1 kirc 11030 lncrna 760
CAT566.1 kirp 11030 lncrna 761
ESAT72.2 embryonic stem cells 7755 lncrna 762
ESAT72.1 embryonic stem cells 8076 lncrna 763
CAT573.1 kich 3527 lncrna 764
CAT573.1 ovarian 3527 lncrna 765
CAT573.2 lusc 3652 lncrna 766
ESAT31.3 embryonic stem cells 3369 tucp 767
ESAT31.4 embryonic stem cells 3469 lncrna 768
ESAT83 embryonic stem cells 319 lncrna 769
CAT576 head neck 450 lncrna 770
CAT576 lusc 450 lncrna 771
CAT577 luad 522 tucp 772
 CAT743 mecmlloblastoma 6575 lncrna 816
CAT743 mecmlloblastoma 6575 lncrna 816
CAT749.1 kirc 10675 lncrna 817
LINC00518.1 melanoma 2922 lncrna 818
LINC00518.7 melanoma 3083 lncrna 819
LINC00518.2 melanoma 3019 lncrna 820
LINC00518.3 melanoma 3188 lncrna 821
LINC00518.6 melanoma 2806 lncrna 822
LINC00518.5 melanoma 3289 lncrna 823
CAT773.1 embryonic stem cells 399 lncrna 824
ZSCAN16-AS1 heart 2914 lncrna 825
ZSCAN16-AS1 kich 2914 lncrna 826
CAT789 mpn 1491 lncrna 827
CAT789 cml 1491 lncrna 828
CAT789 kirc 1491 lncrna 829
CAT789 embryonic stem cells 1491 lncrna 830
CAT789 skeletal muscle 1491 lncrna 831
PRCAT30.2 prostate 5371 lncrna 832
PRCAT30.1 prostate 5396 lncrna 833
PRC AT 15.1 prostate 1663 lncrna 834
PRCAT15.2 prostate 2719 lncrna 835
CAT793.1 igg 9738 lncrna 836
CAT793.1 cervical 9738 lncrna 837
CAT793.1 kirp 9738 lncrna 838
CAT793.1 breast 9738 lncrna 839
CAT793.1 luad 9738 lncrna 840
AMAT92 ami 7843 lncrna 841
CAT800.1 prostate 561 lncrna 842
CAT800.2 prostate 650 lncrna 843
CAT828.1 head neck 5239 lncrna 844
CAT828.1 hiad 5239 lncrna 845
CAT828.1 lusc 5239 lncrna 846
CAT837 cml 3351 lncrna 847
CAT837 breast 3351 lncrna 848
CAT840.1 kich 9492 lncrna 849
CAT840.2 kich 9162 lncrna 850
CAT840.3 embryonic stem cells 5951 lncrna 851
CAT840.4 kich 1448 lncrna 852
CAT840.5 kich 8137 lncrna 853
CMAT40 cml 6761 lncrna 854
KCCAT167 kirc 8975 lncrna 855
CAT862.1 embryonic stem cells 21097 lncrna 856
CAT862.2 embryonic stem cells 602 lncrna 857
CAT862.2 breast 602 lncrna 858
CAT862.3 embryonic stem cells 6909 lncrna 859
CAT870.1 lusc 2047 tucp 860
CAT870.2 gbm 1381 lncrna 861
CAT870.2 lusc 1381 lncrna 862
CAT878.1 prostate 2322 lncrna 863
CAT878.2 prostate 2286 lncrna 864
CAT878.3 ovarian 1126 lncrna 865
CAT878.3 colorectal 1126 lncrna 866
PNAT3 pancreatic 3222 lncrna 867
CAT905 head neck 912 lncrna 868
CAT905 lusc 912 lncrna 869
CAT906 head neck 1465 lncrna 870
CAT906 lusc 1465 lncrna 871
LINC00265.1 kich 5240 tucp 872
CAT932.1 kirp 2798 lncrna 873
CAT932.1 medulloblastoma 2798 lncrna 874
CAT932.2 head neck 2795 lncrna 875
PNAT63.1 pancreatic 1608 lncrna 876
PNAT63.2 pancreatic 1717 lncrna 877
HNCAT59 head neck 3151 lncrna 878
ESAT63.2 embryonic stem cells 1968 lncrna 879
CAT962 embryonic stem cells 3877 lncrna 880
CAT962 kich 3877 lncrna 881
CAT962 luad 3877 lncrna 882
CAT962 lusc 3877 lncrna 883
HRAT17.4 heart 1064 lncrna 884
OVAT20.3 ovarian 695 lncrna 885
OVAT20.4 ovarian 5210 lncrna 886
OVAT20.1 ovarian 4908 lncrna 887
OVAT20.5 ovarian 5017 lncrna 888
CAT972.1 kirc 18548 tucp 889
CAT972.1 kirp 18548 tucp 890
CAT972.1 kirc 18548 tucp 891
CAT972.1 kich 18548 tucp 892
CAT979 kirc 1053 lncrna 893
CAT979 kich 1053 lncrna 894
CAT979 lusc 1053 lncrna 895
CAT989 gbra 1262 tucp 896
CAT989 prostate 1262 tucp 897
ESAT10.4 embryonic stem cells 2160 lncrna 898
ESAT10.1 embryonic stem cells 1879 lncrna 899
ESAT10.2 embryonic stem cells 3150 lncrna 900
ESAT10.3 embryonic stem cells 3206 lncrna 901
 CAT1089.2 melanoma 4663 lncrna 945
CAT1089.2 melanoma 4663 lncrna 945
CAT1089.3 colorectal 676 lncrna 946
CAT1089.3 breast 676 lncrna 947
CAT 1089.3 cml 676 lncrna 948
CAT1089.3 melanoma 676 lncrna 949
CAT1089.3 ami 676 lncrna 950
CAT1089.3 skeletal muscle 676 lncrna 951
CAT1089.3 gbm 676 lncrna 952
CAT1089.3 igg 676 lncrna 953
CAT1089.3 medulloblastoma 676 lncrna 954
CAT1089.3 kirc 676 lncrna 955
THCAT4.1 thyroid 1631 lncrna 956
RNF139-AS1.1 thyroid 15042 lncrna 957
RNF139-AS1.2 head neck 1914 lncrna 958
RNF139-AS1.2 breast 1914 lncrna 959
ESAT82.1 embryonic stem cells 3858 lncrna 960
CAT 1109 head neck 1243 lncrna 961
CAT 1109 skeletal muscle 1243 lncrna 962
CAT 1109 colorectal 1243 lncrna 963
CAT1115.1 lusc 5792 lncrna 964
CAT1115.2 ovarian 1126 lncrna 965
HNCAT56 head neck 621 lncrna 966
FAM83H-AS1.1 prostate 5530 tucp 967
FAM83H-AS1.1 breast 5530 tucp 968
FAM83H-AS1.1 luad 5530 tucp 969
FAM83H-AS1.1 lusc 5530 tucp 970
FAM83H-AS1.1 heart 5530 tucp 971
FAM83H-AS1.1 melanoma 5530 tucp 972
FAM83H-AS1.1 mpn 5530 tucp 973
FAM83H-AS1.1 gbm 5530 tucp 974
FAM83H-AS1.1 igg 5530 tucp 975
FAM83H-AS1.1 medulloblastoma 5530 tucp 976
FAM83H-AS1.2 lusc 6435 tucp 977
FAM83H-AS1.2 gbm 6435 tucp 978
FAM83H-AS1.2 melanoma 6435 tucp 979
FAM83H-AS1.3 gbm 6637 tucp 980
FAM83H-AS1.3 melanoma 6637 tucp 981
FAM83H-AS1.3 medulloblastoma 6637 tucp 982
FAM83H-AS1.4 prostate 3087 lncrna 983
FAM83H-AS1.4 breast 3087 lncrna 984
FAM83H-AS1.4 luad 3087 lncrna 985
FAM83H-AS1.4 lusc 3087 lncrna 986
FAM83H-AS1.4 gbm 3087 lncrna 987
FAM83H-AS1.4 melanoma 3087 lncrna 988
FAM83H-AS1.4 igg 3087 lncrna 989
FAM83H-AS1.4 medulloblastoma 3087 lncrna 990
CAT1129 ami 7255 lncrna 991
CAT1129 embryonic stem cells 7255 lncrna 992
CAT 1129 thyroid 7255 lncrna 993
CAT1129 kirp 7255 lncrna 994
CAT1129 breast 7255 lncrna 995
CAT 1129 luad 7255 lncrna 996
CAT 1129 liver 7255 lncrna 997
CAT1141.1 heart 4172 tucp 998
CAT1141.2 medulloblastoma 4670 tucp 999
CAT1147.1 prostate 1890 lncrna 1000
CAT1147.1 luad 1890 lncrna 1001
CAT1147.1 hisc 1890 lncrna 1002
CAT1147.2 heart 1112 lncrna 1003
CAT1147.2 prostate 1112 lncrna 1004
CAT1147.2 luad 1112 lncrna 1005
CAT1147.2 lusc 1112 lncrna 1006
CAT1147.3 heart 15070 lncrna 1007
CAT1147.3 prostate 15070 lncrna 1008
CAT1147.3 luad 15070 lncrna 1009
CAT1147.3 lusc 15070 lncrna 1010
CAT 1160 head neck 2191 lncrna 1011
CAT 1160 lusc 2191 lncrna 1012
CAT 1162 kirc 1292 lncrna 1013
CAT1162 melanoma 1292 lncrna 1014
CAT1162 thyroid 1292 lncrna 1015
THCAT39.7 thyroid 8539 lncrna 1016
THCAT39.7 thyroid 8539 lncrna 1017
THCAT39.1 thyroid 6408 lncrna 1018
THCAT39.9 thyroid 1265 lncrna 1019
THCAT39.9 thyroid 1265 lncrna 1020
THCAT39.17 thyroid 13258 lncrna 1021
THCAT39.14 thyroid 3327 lncrna 1022
MIR181A2HG.2 thyroid 849 lncrna 1023
MIR181A2HG.1 thyroid 922 lncrna 1024
MIR181A2HG.1 thyroid 922 lncrna 1025
CAT1195.1 kich 10727 lncrna 1026
CAT1195.1 kirp 10727 lncrna 1027
CAT1195.2 kirp 6079 lncrna 1028
CAT1201 cervical 868 lncrna 1029
CAT1201 igg 868 lncrna 1030
CAT1204.1 ovarian 1049 lncrna 1031
CAT1204.1 kirc 1049 lncrna 1032
CAT1204.1 kirp 1049 lncrna 1033
CAT1204.1 prostate 1049 lncrna 1034
CAT1204.1 kich 1049 lncrna 1035
CAT1204.2 cervical 4425 lncrna 1036
CAT1204.3 lusc 1795 lncrna 1037
CAT1204.3 prostate 1795 lncrna 1038
CAT1204.4 lusc 1313 lncrna 1039
CAT1204.4 prostate 1313 lncrna 1040
CAT1212 colorectal 877 lncrna 1041
CAT1212 lusc 877 lncrna 1042
CAT1212 liver 877 lncrna 1043
CAT1212 luad 877 lncrna 1044
CAT1212 prostate 877 lncrna 1045
CAT1212 kich 877 lncrna 1046
CAT1212 breast 877 lncrna 1047
CAT2275.1 prostate 5925 lncrna 1048
CAT2275.2 prostate 2443 lncrna 1049
CAT2275.2 kich 2443 lncrna 1050
CMAT1 cml 3108 lncrna 1051
MBAT1 medulloblastoma 7147 lncrna 1052
ESAT92.2 embryonic stem cells 386 lncrna 1053
ESAT92.1 embryonic stem cells 26348 tucp 1054
ESAT92.3 embryonic stem cells 32499 tucp 1055
MEAT 16 melanoma 662 lncrna 1056
CAT2277.1 melanoma 1373 lncrna 1057
CAT2277.2 uterine 1238 lncrna 1058
CAT2277.3 breast 504 lncrna 1059
CAT2277.3 melanoma 504 lncrna 1060
1061
MP ATI mpn 569 lncrna
1062
ATP6V0E2-AS1.1 thyroid 3969 lncrna
1063
ATP6V0E2-AS1.2 kich 2679 lncrna
1064
ATP6V0E2-AS1.2 thyroid 2679 lncrna
1065
HNCAT1 head neck 1495 lncrna
1066
CAT566.2 kirc 11102 lncrna
1067
CAT264.1 prostate 5674 lncrna
1068
CAT264.1 medulloblastoma 5674 lncrna
1069
CAT264.2 thyroid 5252 lncrna
1070
CAT264.2 kirc 5252 lncrna
1071
CAT264.2 prostate 5252 lncrna
1072
CAT264.2 breast 5252 lncrna
1073
CAT1495.1 thvroid 8833 lncrna
1074
CAT1495.1 kirp 8833 lncrna
1075
CAT1495.2 thyroid 7437 lncrna
1076
CAT1495.3 thvroid 7646 lncrna
1077
CAT1496.1 thyroid 5209 lncrna
1078
CAT1496.1 kirp 5209 lncrna
1079
CAT1496.1 head neck 5209 lncrna
1080
CAT1496.2 thyroid 4778 lncrna
1081
CAT1496.2 kirp 4778 lncrna
1082
CAT1496.3 thyroid 5016 lncrna
CAT1496.3 kirp 5016 lncrna 1083
CAT1496.3 thyroid 5016 lncrna 1084
CAT1496.3 head neck 5016 lncrna 1085
CAT1383.1 kirc 1507 lncrna 1086
CAT1382.1 kirc 1663 lncrna 1087
CAT1382.1 breast 1663 lncrna 1088
CAT1382.2 kirc 930 lncrna 1089
CAT1382.2 breast 930 lncrna 1090
GBAT2 gbm 666 lncrna 1091
KCCAT148.1 kirc 2950 lncrna 1092
KCCAT148.1 kirc 2950 lncrna 1093
CAT329 kirc 1343 lncrna 1094
CAT329 luad 1343 lncrna 1095
CAT329 lusc 1343 lncrna 1096
LINC00511.4 lusc 12087 lncrna 1097
SBF2-AS1.2 kich 955 lncrna 1098
SBF2-AS1.3 kich 1210 lncrna 1099
SBF2-AS1.4 liver 1216 lncrna 1100
SBF2-AS1.4 luad 1216 lncrna 1101
SBF2-AS1.4 lusc 1216 lncrna 1102
VCAN-AS1 pancreatic 522 lncrna 1103
VCAN-AS1 kirp 522 lncrna 1104
VCAN-AS1 stomach 522 lncrna 1105
VCAN-AS1 breast 522 lncrna 1106
GATA3-AS1.3 breast 1059 lncrna 1107
GATA3-AS1.3 breast 1059 lncrna 1108
GATA3-AS1.3 kirc 1059 lncrna 1109
GATA3-AS1.3 prostate 1059 lncrna 1110
GATA3-AS1.4 breast 4620 lncrna 1111
GATA3-AS1.4 kirc 4620 lncrna 1112
GATA3-AS1.5 breast 4613 lncrna 1113
GATA3-AS1.5 breast 4613 lncrna 1114
GATA3-AS1.5 prostate 4613 lncrna 1115
CAT800.3 kich 638 lncrna 1116
HRAT70 heart 674 lncrna 1117
CAT162.1 skeletal muscle 8558 lncrna 1118
CAT162.1 ami 8558 lncrna 1119
CAT162.1 colorectal 8558 lncrna 1120
CAT162.1 prostate 8558 lncrna 1121
CAT162.1 kich 8558 lncrna 1122
CAT162.1 luad 8558 lncrna 1123
CAT162.1 breast 8558 lncrna 1124
CAT162.2 skeletal muscle 8749 lncrna 1125
CAT162.2 cml 8749 lncrna 1126
CAT162.2 prostate 8749 lncrna 1127
CAT162.2 kich 8749 lncrna 1128
CAT162.2 luad 8749 lncrna 1129
CAT162.2 breast 8749 lncrna 1130
CAT1852.1 thyroid 4821 lncrna 1131
CAT1852.2 thyroid 9499 tucp 1132
CAT1852.2 head neck 9499 tucp 1133
CAT1852.2 liver 9499 tucp 1134
CAT1852.2 kirc 9499 tucp 1135
CAT1852.2 prostate 9499 tucp 1136
CAT1852.2 kich 9499 tucp 1137
CAT1852.2 breast 9499 tucp 1138
LINC00545 embryonic stem cells 677 lncrna 1139
CAT664.2 kich 4397 lncrna 1140
CAT2064 thyroid 2859 lncrna 1141
CAT2064 kirp 2859 lncrna 1142
CAT2064 prostate 2859 lncrna 1143
CAT2064 breast 2859 lncrna 1144
CAT2064 gbm 2859 lncrna 1145
CAT2064 igg 2859 lncrna 1146
CAT2064 skeletal muscle 2859 lncrna 1147
CAT1591 ami 16567 lncrna 1148
CAT1591 kirc 16567 lncrna 1149
CAT793.2 ovarian 8428 lncrna 1150
THCAT36.1 thyroid 1195 lncrna 1151
THCAT36.1 thyroid 1195 lncrna 1152
MIR205HG.4 lusc 2515 lncrna 1153
MIR205HG.4 gbm 2515 lncrna 1154
MIR205HG.4 prostate 2515 lncrna 1155
MIR205HG.4 breast 2515 lncrna 1156
THCAT36.4 thyroid 1355 lncrna 1157
THCAT36.4 thyroid 1355 lncrna 1158
MIR205HG.5 lusc 2829 lncrna 1159
CAT969.1 thyroid 1371 lncrna 1160
MIR205HG.6 lusc 3963 lncrna 1161
CAT1664 thyroid 605 lncrna 1162
CAT1664 lusc 605 lncrna 1163
CAT 1664 kirp 605 lncrna 1164
CAT 1664 luad 605 lncrna 1165
CAT 1664 kirc 605 lncrna 1166
CAT 1664 kich 605 lncrna 1167
SMAT24 skeletal muscle 531 lncrna 1168
CAT2176.2 breast 2212 lncrna 1169
CAT2176.3 breast 671 lncrna 1170
CAT2176.3 breast 671 lncrna 1171
CAT2157 head neck 2017 lncrna 1172
CAT2157 lusc 2017 lncrna 1173
CAT1546 kirc 860 lncrna 1174
CAT1546 kirp 860 lncrna 1175
MY016-AS1 luad 4439 lncrna 1176
WT1-AS.4 ovarian 10678 lncrna 1177
WT1-AS.5 ovarian 9901 lncrna 1178
WT1-AS.6 ovarian 10532 lncrna 1179
LINC00087 kich 5585 tucp 1180
LINC00087 colorectal 5585 tucp 1181
LINC00087 thyroid 5585 tucp 1182
LINC00087 kirp 5585 tucp 1183
LINC00087 bladder 5585 tucp 1184
LINC00087 breast 5585 tucp 1185
LINC00087 luad 5585 tucp 1186
LINC00087 kirc 5585 tucp 1187
LINC00087 prostate 5585 tucp 1188
LINC00152.3 thyroid 3019 lncrna 1189
LINC00152.3 kirc 3019 lncrna 1190
LINC00152.3 liver 3019 lncrna 1191
LINC00152.3 luad 3019 lncrna 1192
ESAT15.2 embryonic stem cells 1906 lncrna 1193
LSCAT1.2 lusc 6456 tucp 1194
CAT171.2 stomach 36752 lncrna 1195
CAT171.3 thyroid 6660 lncrna 1196
THCAT39.3 thyroid 695 lncrna 1197
THCAT39.3 thyroid 695 lncrna 1198
THCAT39.i l thyroid 1445 lncrna 1199
CAT2071 mpn 4868 lncrna 1200
CAT2071 colorectal 4868 lncrna 1201
CAT2071 kirc 4868 lncrna 1202
CAT2071 luad 4868 lncrna 1203
CAT2071 lusc 4868 lncrna 1204
PRCAT102.2 prostate 667 lncrna 1205
ME AT 1.1 melanoma 1700 lncrna 1206
CAT1325 kich 6235 lncrna 1207
CAT 1325 medulloblastoma 6235 lncrna 1208
CAT 1947.1 head neck 2165 lncrna 1209
CAT 1947.1 prostate 2165 lncrna 1210
CAT1947.2 head neck 3123 tucp 1211
CAT742.4 kirp 4914 lncrna 1212
CAT742.5 kich 4909 lncrna 1213
CAT742.5 kich 4909 lncrna 1214
HNCAT25.1 head neck 1733 lncrna 1215
HNCAT25.3 head neck 1168 lncrna 1216
HNCAT25.2 head neck 784 lncrna 1217
PRCAT101 prostate 2254 lncrna 1218
GBAT18 gbm 2013 lncrna 1219
THCAT3 thyroid 2016 lncrna 1220
CAT1768.2 prostate 1209 lncrna 1221
TRPC7-AS1 pancreatic 5009 lncrna 1222
CAT828.2 head neck 15379 lncrna 1223
CAT828.2 luad 15379 lncrna 1224
CAT828.2 lusc 15379 lncrna 1225
CAT1284.2 kich 5397 lncrna 1226
CAT1284.3 kirc 2413 lncrna 1227
CAT1284.3 lusc 2413 lncrna 1228
CAT1284.3 pancreatic 2413 lncrna 1229
CAT1284.3 prostate 2413 lncrna 1230
LBX2-AS1.1 head neck 4528 lncrna 1231
LBX2-AS1.1 breast 4528 lncrna 1232
LBX2-AS1.1 kirc 4528 lncrna 1233
LBX2-AS1.2 thyroid 1086 lncrna 1234
LBX2-AS1.2 kirc 1086 lncrna 1235
LBX2-AS1.2 kirp 1086 lncrna 1236
LBX2-AS1.2 luad 1086 lncrna 1237
ESAT51 embryonic stem cells 7090 lncrna 1238
CAT2176.4 breast 2564 lncrna 1239
CAT2176.4 breast 2564 lncrna 1240
CAT 1946 uterine 700 lncrna 1241
CAT 1946 ovarian 700 lncrna 1242
ESAT4 embryonic stem cells 1072 lncrna 1243
HRAT1 heart 323 lncrna 1244
PNAT13.2 pancreatic 1430 lncrna 1245
CAT1489 head neck 277 lncrna 1246
CAT1489 lusc 277 lncrna 1247
CAT682.2 breast 4292 lncrna 1248
CAT682.2 luad 4292 lncrna 1249
CAT682.2 breast 4292 lncrna 1250
CAT682.2 kirc 4292 lncrna 1251
CAT682.2 kirp 4292 lncrna 1252
CAT682.2 kich 4292 lncrna 1253
LSCAT1.5 lusc 4306 lncrna 1254
GBAT8 gbm 1276 lncrna 1255
CAT1919 uterine 655 lncrna 1256
CAT1919 luad 655 lncrna 1257
ESAT31.2 embryonic stem cells 5275 lncrna 1258
ESAT31.5 embryonic stem cells 5977 lncrna 1259
ESAT31.1 embryonic stem cells 6396 lncrna 1260
CAT1858 colorectal 567 lncrna 1261
CAT1858 kirc 567 lncrna 1262
CAT1393 colorectal 816 lncrna 1263
CAT 1393 kirc 816 lncrna 1264
CAT 1393 skeletal muscle 816 lncrna 1265
CAT 1393 prostate 816 lncrna 1266
CMAT2 cml 1201 lncrna 1267
CAT1957.2 head neck 494 lncrna 1268
CAT1957.2 breast 494 lncrna 1269
CAT1957.2 luad 494 lncrna 1270
CAT1957.2 lusc 494 lncrna 1271
CMAT28 cml 1214 lncrna 1272
CAT1195.3 kich 8390 lncrna 1273
CAT2186.2 head neck 4333 lncrna 1274
PRCAT104.7 prostate 7804 tucp 1275
PRCAT104.7 prostate 7804 tucp 1276
PRCAT104.1 prostate 7781 tucp 1277
PRCAT104.1 prostate 7781 tucp 1278
CAT2227 prostate 9019 lncrna 1279
CAT2227 prostate 9019 lncrna 1280
CAT2227 thyroid 9019 lncrna 1281
PRCAT104.2 prostate 4575 lncrna 1282
PRCAT104.2 prostate 4575 lncrna 1283
PRCAT104.5 prostate 10052 tucp 1284
PRCAT104.5 prostate 10052 tucp 1285
UTAT18 uterine 557 lncrna 1286
MIR205HG.7 lusc 2018 lncrna 1287
MIR205HG.7 prostate 2018 lncrna 1288
CAT1472.2 kirp 6055 lncrna 1289
CAT1472.2 lusc 6055 lncrna 1290
CAT1897 uterine 622 lncrna 1291
CAT1897 kirc 622 lncrna 1292
CAT366.2 thyroid 1512 lncrna 1293
CAT366.3 thyroid 2528 lncrna 1294
CAT366.3 stomach 2528 lncrna 1295
CAT366.3 lusc 2528 lncrna 1296
CAT366.3 head neck 2528 lncrna 1297
CAT366.3 breast 2528 lncrna 1298
CAT366.3 luad 2528 lncrna 1299
THCAT39.4 thyroid 1747 lncrna 1300
THCAT39.4 thyroid 1747 lncrna 1301
THCAT39.6 thyroid 18125 lncrna 1302
THCAT39.6 thyroid 18125 lncrna 1303
THCAT39.13 thyroid 1507 lncrna 1304
THCAT39.13 thyroid 1507 lncrna 1305
THCAT39.8 thyroid 8126 lncrna 1306
THCAT39.8 thyroid 8126 lncrna 1307
THCAT39.16 thyroid 1140 lncrna 1308
THCAT39.12 thyroid 1375 lncrna 1309
UTAT40 uterine 4336 tucp 1310
UTAT2 uterine 535 lncrna 1311
CAT1023.1 breast 1144 lncrna 1312
CAT1023.1 lusc 1144 lncrna 1313
TTN-AS1.4 heart 1146 lncrna 1314
CAT612.1 lusc 5363 lncrna 1315
CAT612.2 head neck 9926 tucp 1316
CAT612.2 lusc 9926 tucp 1317
CAT932.3 stomach 4148 lncrna 1318
CAT2267 head neck 2709 lncrna 1319
CAT2267 kirp 2709 lncrna 1320
CAT2267 breast 2709 lncrna 1321
CAT2267 luad 2709 lncrna 1322
CAT2267 kirc 2709 lncrna 1323
HRAT40.1 heart 3737 lncrna 1324
HRAT40.2 heart 4209 lncrna 1325
ESAT63.3 embrvonic stem cells 3502 lncrna 1326
ESAT63.1 embryonic stem cells 996 lncrna 1327
LINC00678.6 embryonic stem cells 5446 lncrna 1328
CAT 1860 cml 4554 lncrna 1329
CAT 1860 kirc 4554 lncrna 1330
CAT 1860 kich 4554 lncrna 1331
CAT229 kirc 1780 lncrna 1332
CAT229 kirc 1780 lncrna 1333
CAT229 kich 1780 lncrna 1334
CAT2268 prostate 6181 lncrna 1335
CAT2268 cervical 6181 lncrna 1336
CAT2268 ovarian 6181 lncrna 1337
CAT 1370 breast 642 lncrna 1338
CAT 1370 luad 642 lncrna 1339
CAT 1370 lusc 642 lncrna 1340
PCAT6 luad 1849 lncrna 1341
PCAT6 lusc 1849 lncrna 1342
CAT152.1 thyroid 1963 lncrna 1343
CAT152.1 breast 1963 lncrna 1344
CAT152.1 luad 1963 lncrna 1345
CAT152.1 lusc 1963 lncrna 1346
CAT152.2 kirp 2167 lncrna 1347
CAT152.2 prostate 2167 lncrna 1348
CAT152.2 breast 2167 lncrna 1349
CAT152.2 luad 2167 lncrna 1350
CAT152.2 lusc 2167 lncrna 1351
CAT76.1 kirc 1119 lncrna 1352
CAT2067 uterine 1391 lncrna 1353
CAT2067 head neck 1391 lncrna 1354
CAT2067 breast 1391 lncrna 1355
CAT2067 medulloblastoma 1391 lncrna 1356
HNCAT13 head neck 1471 lncrna 1357
MIR22HG.1 mpn 2041 lncrna 1358
MIR22HG.1 heart 2041 lncrna 1359
MIR22HG.1 embryonic stem cells 2041 lncrna 1360
MIR22HG.1 thyroid 2041 lncrna 1361
MIR22HG.1 stomach 2041 lncrna 1362
MIR22HG.1 lusc 2041 lncrna 1363
MIR22HG.1 kirp 2041 lncrna 1364
MIR22HG.1 bladder 2041 lncrna 1365
MIR22HG.1 liver 2041 lncrna 1366
MIR22HG.1 luad 2041 lncrna 1367
MIR22HG.1 prostate 2041 lncrna 1368
MIR22HG.1 breast 2041 lncrna 1369
MIR22HG.2 prostate 2158 lncrna 1370
KCCAT40 kirc 56307 lncrna 1371
ESAT13.2 embryonic stem cells 515 lncrna 1372
SNHG12.4 kirc 1934 lncrna 1373
CAT2082.2 thyroid 3487 lncrna 1374
CAT342.1 prostate 1241 lncrna 1375
CAT342.2 lusc 107308 lncrna 1376
THCAT39.2 thyroid 1199 lncrna 1377
THCAT39.2 thyroid 1199 lncrna 1378
THCAT39.15 thyroid 5673 lncrna 1379
LINC00518.4 melanoma 2704 lncrna 1380
ESAT66 embryonic stem cells 11196 lncrna 1381
LINC00371.1 embryonic stem cells 823 tucp 1382
LINC00371.2 embryonic stem cells 1126 lncrna 1383
TTN-AS1.5 heart 1744 lncrna 1384
CAT2039.1 breast 1624 lncrna 1385
BRCAT19 breast 4385 tucp 1386
CAT2039.2 breast 1464 lncrna 1387 CCAT4 kirc 5015 lncrna 1388
CAT1967.2 thyroid 1099 lncrna 1389
CAT1967.2 prostate 1099 lncrna 1390
CAT655.1 head neck 7282 lncrna 1391
CAT655.1 kirc 7282 lncrna 1392
CAT655.2 head neck 21099 lncrna 1393
CAT655.2 kirp 21099 lncrna 1394
CAT655.2 liiad 21099 lncrna 1395
MIR7-3HG.12 pancreatic 3802 lncrna 1396
MIR7-3HG.21 pancreatic 1964 lncrna 1397
MIR7-3HG.8 pancreatic 1967 lncrna 1398
MIR7-3HG.4 pancreatic 1627 lncrna 1399
MIR7-3HG.5 pancreatic 1572 lncrna 1400
MIR7-3HG.13 pancreatic 3273 lncrna 1401
MEAT 1.3 melanoma 15524 tucp 1402
ME AT 1.2 melanoma 1006 lncrna 1403
UTAT51.2 uterine 3938 lncrna 1404
CAT2218 kirp 630 lncrna 1405
CAT2218 medulloblastoma 630 lncrna 1406
MBAT8 medulloblastoma 2035 lncrna 1407
CAT313.6 kirc 3210 lncrna 1408
CAT313.7 kirc 2984 lncrna 1409
MEAT20.4 melanoma 875 lncrna 1410
KHCAT98 kich 2408 lncrna 1411
THCAT39.5 thyroid 6854 lncrna 1412
THCAT39.5 thvroid 6854 lncrna 1413
CAT1272 mpn 8184 lncrna 1414
CAT1272 cml 8184 lncrna 1415
CAT1272 kirc 8184 lncrna 1416
CAT1272 breast 8184 lncrna 1417
CAT773.2 breast 1293 lncrna 1418
CAT773.2 breast 1293 lncrna 1419
CAT868 thyroid 1970 lncrna 1420
CAT868 lusc 1970 lncrna 1421
CAT868 gbm 1970 lncrna 1422
CAT868 igg 1970 lncrna 1423
CAT868 prostate 1970 lncrna 1424
CAT868 liver 1970 lncrna 1425
CAT1485 kirc 1501 lncrna 1426
CAT1485 mpn 1501 lncrna 1427
CAT1485 crnl 1501 lncrna 1428
CAT1485 embryonic stem cells 1501 lncrna 1429
CAT1485 medulloblastoma 1501 lncrna 1430
CAT1485 prostate 1501 lncrna 1431
CAT1485 breast 1501 lncrna 1432
CAT 1580 ovarian 515 lncrna 1433
CAT 1580 medulloblastoma 515 lncrna 1434
ESAT37 embryonic stem cells 4865 lncrna 1435
LINC00668.3 head neck 2625 lncrna 1436
LINC00668.3 lusc 2625 lncrna 1437
KCCAT200 kirc 2339 lncrna 1438
CAT 1828 kirc 2309 lncrna 1439
CAT 1828 prostate 2309 lncrna 1440
CAT1828 breast 2309 lncrna 1441
CAT 1828 luad 2309 lncrna 1442
MEAT48.2 melanoma 24636 lncrna 1443
MEAT48.3 melanoma 24396 lncrna 1444
CAT969.2 breast 3312 lncrna 1445
CAT969.2 luad 3312 lncrna 1446
KCCAT131.1 kirc 2547 lncrna 1447 CCAT131.1 kirc 2547 lncrna 1448
KCCAT131.3 kirc 2221 lncrna 1449
KCCAT131.2 kirc 3651 lncrna 1450
KCCAT131.2 kirc 3651 lncrna 1451
CAT1113.1 colorectal 1852 lncrna 1452
FAM83H-AS1.5 prostate 10522 tucp 1453
FAM83H-AS1.5 breast 10522 tucp 1454
FAM83H-AS1.5 luad 10522 tucp 1455
FAM83H-AS1.5 lusc 10522 tucp 1456
FAM83H-AS1.5 heart 10522 tucp 1457
FAM83H-AS1.5 gbm 10522 tucp 1458
FAM83H-AS1.5 melanoma 10522 tucp 1459
LINC01003 gbm 3779 lncrna 1460
CAT1023.2 breast 13105 lncrna 1461
CAT1456 colorectal 1916 lncrna 1462
CAT1456 uterine 1916 lncrna 1463
CAT1456 breast 1916 lncrna 1464
CAT1456 lusc 1916 lncrna 1465
THCAT36.9 thyroid 1259 lncrna 1466
CAT1576 kirc 1579 lncrna 1467
CAT1576 medulloblastoma 1579 lncrna 1468
MIR7-3HG.14 pancreatic 3421 lncrna 1469
MIR7-3HG.i l pancreatic 3252 lncrna 1470
MIR7-3HG.15 pancreatic 3304 lncrna 1471
MIR7-3HG.17 pancreatic 3560 lncrna 1472
MIR7-3HG.9 pancreatic 3491 lncrna 1473
ESAT39.1 embrvonic stem cells 3310 lncrna 1474
ESAT39.2 embryonic stem cells 12934 tucp 1475
MIR4435-1HG.3 breast 903 lncrna 1476
MIR4435-1HG.3 luad 903 lncrna 1477
MIR4435-1HG.3 liver 903 lncrna 1478
MIR4435-1HG.3 igg 903 lncrna 1479
CAT271 luad 1212 lncrna 1480
CAT271 lusc 1212 lncrna 1481
MEAT51.2 melanoma 2746 lncrna 1482
MEAT51.1 melanoma 2650 lncrna 1483
CAT249.2 embryonic stem cells 11296 tucp 1484
CAT249.2 kich 11296 tucp 1485
BRCAT24.2 breast 1938 lncrna 1486
CAT2164.1 prostate 5670 lncrna 1487
CAT2164.1 breast 5670 lncrna 1488
CAT2164.1 luad 5670 lncrna 1489
CAT2164.2 heart 3981 lncrna 1490
CAT2164.2 cml 3981 lncrna 1491
CAT2164.2 ami 3981 lncrna 1492
CAT2164.2 igg 3981 lncrna 1493
CAT2164.2 lusc 3981 lncrna 1494
CAT2164.2 prostate 3981 lncrna 1495
CAT2164.2 kich 3981 lncrna 1496
CAT2164.2 luad 3981 lncrna 1497
CAT2164.2 breast 3981 lncrna 1498
KPCAT19 kirp 632 lncrna 1499
MEAT38 melanoma 568 lncrna 1500
MIR31HG.1 thyroid 2183 lncrna 1501
MIR31HG.1 prostate 2183 lncrna 1502
MIR31HG.2 thyroid 2442 lncrna 1503
MIR31HG.2 prostate 2442 lncrna 1504
THCAT22.2 thyroid 7479 lncrna 1505
THCAT22.2 thvroid 7479 lncrna 1506
THCAT22.5 thyroid 3138 lncrna 1507
THCAT22.5 thyroid 3138 lncrna 1508
CAT226.7 melanoma 1278 lncrna 1509
CAT226.8 melanoma 599 lncrna 1510
LINC00665.6 ovarian 5220 lncrna 1511
LINC00665.6 prostate 5220 lncrna 1512
LINC00665.6 breast 5220 lncrna 1513
LINC00665.6 luad 5220 lncrna 1514
LINC00665.6 lusc 5220 lncrna 1515
LINC00665.6 colorectal 5220 lncrna 1516
LINC00665.7 breast 5165 lncrna 1517
CAT944 ovarian 553 lncrna 1518
CAT944 colorectal 553 lncrna 1519
CAT944 uterine 553 lncrna 1520
CAT944 breast 553 lncrna 1521
CAT944 melanoma 553 lncrna 1522
LINC00665.8 ovarian 3759 lncrna 1523
LINC00665.8 prostate 3759 lncrna 1524
LINC00665.8 breast 3759 lncrna 1525
LINC00665.8 luad 3759 lncrna 1526
LINC00665.8 lusc 3759 lncrna 1527
LINC00665.8 colorectal 3759 lncrna 1528
DDX11-AS1.2 lusc 3230 lncrna 1529
KCCAT162 kirc 16275 lncrna 1530
CAT2251.2 mpn 2297 lncrna 1531
CAT2251.2 ami 2297 lncrna 1532
LINC00265.3 kich 4722 tucp 1533
LINC00265.2 kich 4851 tucp 1534
CAT 1326 ami 1076 lncrna 1535
CAT 1326 kirc 1076 lncrna 1536
CAT 1326 lusc 1076 lncrna 1537
CAT529 thyroid 1995 lncrna 1538
CAT529 lusc 1995 lncrna 1539
THCAT57 thvroid 938 lncrna 1540
CAT1683.2 thvroid 5074 lncrna 1541
CAT1683.2 lusc 5074 lncrna 1542
CAT1683.2 kirp 5074 lncrna 1543
CAT1683.2 bladder 5074 lncrna 1544
CAT1683.2 luad 5074 lncrna 1545
CAT1683.2 kirc 5074 lncrna 1546
CAT1345.2 luad 822 lncrna 1547
CAT1345.2 lusc 822 lncrna 1548
KCCAT279.2 kirc 2100 lncrna 1549 CCAT279.1 kirc 2224 lncrna 1550 CCAT279.1 kirc 2224 lncrna 1551
BRCAT24.1 breast 1593 lncrna 1552
BRCAT24.1 breast 1593 lncrna 1553
BRCAT24.3 breast 2882 lncrna 1554
MPAT2 mpn 6029 lncrna 1555
CAT1435 liver 404 lncrna 1556
CAT1435 mpn 404 lncrna 1557
CAT1435 cml 404 lncrna 1558
CAT1435 ami 404 lncrna 1559
CAT1435 kirp 404 lncrna 1560
CAT1435 kich 404 lncrna 1561
CAT226.9 kirc 1604 lncrna 1562
CAT99.3 colorectal 4273 lncrna 1563
CAT99.3 uterine 4273 lncrna 1564
CAT99.3 kich 4273 lncrna 1565
CAT99.3 luad 4273 lncrna 1566
CAT99.3 lusc 4273 lncrna 1567
ESAT33.3 embryonic stem cells 78505 lncrna 1568
MIR31HG.3 thyroid 4972 lncrna 1569
CAT 1807 kich 1795 lncrna 1570
CAT 1807 breast 1795 lncrna 1571
HOXA11-AS.1 head neck 9419 lncrna 1572
HOXA11-AS.1 lusc 9419 lncrna 1573
HOXA11-AS.2 kirc 7671 lncrna 1574
HOXA11-AS.3 lusc 7195 tucp 1575
CAT1768.3 prostate 5769 lncrna 1576
CAT1768.3 luad 5769 lncrna 1577
CAT1768.4 prostate 4744 lncrna 1578
PRCAT121.2 prostate 2141 lncrna 1579
PRCAT121.1 prostate 6441 lncrna 1580
CAT359.2 kich 1706 lncrna 1581
CAT359.2 kich 1706 lncrna 1582
CAT359.3 kich 3119 lncrna 1583
CAT359.3 kich 3119 lncrna 1584
CAT359.3 kirp 3119 lncrna 1585
LIMD1-AS1.1 kich 1252 lncrna 1586
LIMD1-AS1.1 luad 1252 lncrna 1587
LIMD1-AS1.1 lusc 1252 lncrna 1588
CAT226.10 kich 12650 lncrna 1589
CAT227.4 kirc 11959 lncrna 1590
CAT227.4 kirc 11959 lncrna 1591
CAT227.5 kirc 2100 lncrna 1592
CAT227.5 kirc 2100 lncrna 1593
CAT226.11 kich 28708 lncrna 1594
CAT227.6 kirc 14049 lncrna 1595
CAT474.2 embryonic stem cells 18915 tucp 1596
CAT474.3 ovarian 11888 lncrna 1597
ESAT9 embryonic stem cells 1048 lncrna 1598
CMAT7 cml 724 lncrna 1599
CAT1532 gbm 3711 lncrna 1600
CAT1532 igg 3711 lncrna 1601
CAT1532 kirp 3711 lncrna 1602
CAT1532 liver 3711 lncrna 1603
CAT1532 colorectal 3711 lncrna 1604
CAT1532 thyroid 3711 lncrna 1605
AMAT47 ami 1525 tucp 1606
CAT1439 medulloblastoma 770 lncrna 1607
CAT1439 kirc 770 lncrna 1608
CAT1439 mpn 770 lncrna 1609
CAT1439 cml 770 lncrna 1610
CAT1439 thyroid 770 lncrna 1611
CAT1439 head neck 770 lncrna 1612
ESRG.6 ovarian 10002 lncrna 1613
GBAT14 gbm 1042 lncrna 1614
KHCAT3.2 kich 1083 lncrna 1615
GBAT5 gbm 459 lncrna 1616
CAT1736.2 luad 2444 lncrna 1617
TINCR.l thyroid 4657 tucp 1618
TINCR.l breast 4657 tucp 1619
TINCR.l kirc 4657 tucp 1620
TINCR.l luad 4657 tucp 1621
THCAT63 thyroid 2818 tucp 1622
KHCAT1 kich 609 lncrna 1623
AFAP1-AS1 luad 6729 lncrna 1624
PRCAT47.3 prostate 2543 lncrna 1625
PRCAT47.3 prostate 2543 lncrna 1626
PRCAT47.2 prostate 2261 lncrna 1627
PRCAT47.2 prostate 2261 lncrna 1628
PRCAT47.4 prostate 2375 lncrna 1629
PRCAT47.4 prostate 2375 lncrna 1630
WT1-AS.7 ovarian 2477 lncrna 1631
THCAT4.2 thyroid 1860 lncrna 1632
CAT252.2 breast 11636 lncrna 1633
CAT252.2 luad 11636 lncrna 1634
CAT742.6 kich 4836 lncrna 1635
CAT742.6 kich 4836 lncrna 1636
CAT788 ami 837 lncrna 1637
CAT788 kirc 837 lncrna 1638
LSCAT5 lusc 1505 lncrna 1639
IDI2-AS1 melanoma 615 lncrna 1640
IDI2-AS1 head neck 615 lncrna 1641
KCCAT6 kirc 2565 lncrna 1642
KCCAT6 kirc 2565 lncrna 1643
GBAT25.2 gbm 6794 lncrna 1644
CAT2062 thvroid 1317 lncrna 1645
CAT2062 kirc 1317 lncrna 1646
CAT2062 lusc 1317 lncrna 1647
ESAT82.2 embryonic stem cells 3839 lncrna 1648
PNAT1.1 pancreatic 863 lncrna 1649
ESAT1.2 embryonic stem cells 1686 lncrna 1650
ESAT1.1 embryonic stem cells 1196 lncrna 1651
TUG1.4 head neck 5572 lncrna 1652
PN ATI 3.1 pancreatic 1846 lncrna 1653
LI C00152.4 kirc 2305 lncrna 1654
LINC00152.4 stomach 2305 lncrna 1655
LINC00958.6 lusc 38472 lncrna 1656
LINC00958.6 pancreatic 38472 lncrna 1657
LINC00958.7 lusc 22404 lncrna 1658
LINC00958.7 pancreatic 22404 lncrna 1659
LINC00958.7 prostate 22404 lncrna 1660
LINC00958.8 lusc 38382 lncrna 1661
LINC00958.8 pancreatic 38382 lncrna 1662
LINC00958.9 thyroid 16426 lncrna 1663
LINC00958.9 head neck 16426 lncrna 1664
LINC00958.9 lusc 16426 lncrna 1665
LINC00958.9 pancreatic 16426 lncrna 1666
LINC00958.9 prostate 16426 lncrna 1667
CAT1012.2 thyroid 2674 lncrna 1668
CAT1012.2 thyroid 2674 lncrna 1669
CAT 1846 colorectal 1948 lncrna 1670
CAT1846 uterine 1948 lncrna 1671
CAT1381 head neck 6977 lncrna 1672
CAT1381 stomach 6977 lncrna 1673
CAT1381 lusc 6977 lncrna 1674
CAT1768.5 prostate 2715 lncrna 1675
CAT1768.5 luad 2715 lncrna 1676
CAT1768.5 cervical 2715 lncrna 1677
CAT1768.5 melanoma 2715 lncrna 1678
CAT1768.5 kirp 2715 lncrna 1679
CAT1768.6 prostate 16798 lncrna 1680
CAT1575.1 colorectal 1254 lncrna 1681
CAT1575.1 cervical 1254 lncrna 1682
CAT1575.2 kich 2362 lncrna 1683
CAT1575.2 thyroid 2362 lncrna 1684
CAT591 gbm 4547 lncrna 1685
CAT591 igg 4547 lncrna 1686
CAT591 kirc 4547 lncrna 1687
CAT591 kich 4547 lncrna 1688
CAT2082.3 thyroid 11813 tucp 1689
OVAT92 ovarian 2661 lncrna 1690
OVAT131 ovarian 2875 lncrna 1691
CAT2052 breast 891 lncrna 1692
CAT2052 heart 891 lncrna 1693
CAT2052 melanoma 891 lncrna 1694
CAT2052 ami 891 lncrna 1695
CAT2052 skeletal muscle 891 lncrna 1696
CAT2052 gbm 891 lncrna 1697
CAT2052 igg 891 lncrna 1698
CAT2052 medulloblastoma 891 lncrna 1699
CAT2052 head neck 891 lncrna 1700
LACAT23 luad 2080 lncrna 1701
CAT1363.3 uterine 5104 lncma 1702
CAT2168.1 breast 4445 lncrna 1703
CAT227.7 kirc 2301 lncrna 1704
CAT227.7 kirc 2301 lncrna 1705
ESAT32.1 embryonic stem cells 3723 lncrna 1706
CAT969.3 thyroid 871 lncrna 1707
CAT458 ami 13405 lncrna 1708
CAT458 breast 13405 lncrna 1709
PRCAT47.1 prostate 11599 lncrna 1710
PRCAT47.1 prostate 11599 lncrna 1711
CAT260.1 kirc 7684 lncrna 1712
LINC00938 medulloblastoma 2932 lncrna 1713
LINC00938 head neck 2932 lncrna 1714
HNCAT39.2 head neck 792 lncrna 1715
HNCAT39.1 head neck 489 lncrna 1716
CAT1137 ami 734 lncrna 1717
CAT1137 kirc 734 lncrna 1718
CAT1137 gbm 734 lncrna 1719
CAT1137 ovarian 734 lncrna 1720
EPB41L4A-AS1.2 igg 1557 lncrna 1721
EPB41L4A-AS1.2 head neck 1557 lncrna 1722
EPB41L4A-AS1.2 breast 1557 lncrna 1723
EPB41L4A-AS1.3 kirc 856 lncrna 1724
EPB41L4A-AS1.3 head neck 856 lncrna 1725
CAT715 igg 1872 lncrna 1726
CAT715 uterine 1872 lncrna 1727
GBAT25.3 gbm 5608 lncrna 1728
CAT1237.1 liver 23796 lncrna 1729
CAT1237.1 pancreatic 23796 lncrna 1730
CAT1237.2 prostate 1508 lncrna 1731
ST8SIA6-AS1.3 prostate 10062 lncrna 1732
ST8SIA6-AS1.3 liver 10062 lncrna 1733
ST8SIA6-AS1.4 liver 7702 lncrna 1734
ST8SIA6-AS1.5 prostate 6276 lncrna 1735
ST8SIA6-AS1.5 liver 6276 lncrna 1736
LINC00678.3 embryonic stem cells 5513 lncrna 1737
LINC00678.2 embryonic stem cells 5593 lncrna 1738
LINC00678.5 embryonic stem cells 5362 lncrna 1739
LINC00678.4 embryonic stem cells 5517 lncrna 1740
SNHG12.5 kirc 1470 lncrna 1741
CAT2039.3 prostate 1813 lncrna 1742
CAT2039.3 breast 1813 lncrna 1743
CAT2039.3 luad 1813 lncrna 1744
CAT2180.3 kirc 2859 lncrna 1745
CAT2180.3 kirp 2859 lncrna 1746
CAT2180.3 stomach 2859 lncrna 1747
CAT2180.3 medulloblastoma 2859 lncrna 1748
CAT2180.3 luad 2859 lncrna 1749
CAT2180.3 lusc 2859 lncrna 1750
CAT2180.4 cml 3044 lncrna 1751
CAT2180.4 liver 3044 lncrna 1752
CAT2180.4 luad 3044 lncrna 1753
CAT2180.4 lusc 3044 lncrna 1754
ESAT75 embryonic stem cells 4294 lncrna 1755
CAT 184 head neck 1098 lncrna 1756
CAT184 luad 1098 lncrna 1757
CAT821.1 ovarian 9000 lncrna 1758
CAT821.1 colorectal 9000 lncrna 1759
CAT821.1 uterine 9000 lncrna 1760
CAT821.1 head neck 9000 lncrna 1761
CAT821.1 breast 9000 lncrna 1762
CAT821.2 cml 5562 lncrna 1763
CAT821.2 igg 5562 lncrna 1764
CAT821.2 ovarian 5562 lncrna 1765
CAT821.2 head neck 5562 lncrna 1766
CAT821.2 prostate 5562 lncrna 1767
CAT821.2 breast 5562 lncrna 1768
CAT821.2 luad 5562 lncrna 1769
BRCAT1.6 breast 27927 lncrna 1770
BRCAT1.6 breast 27927 lncrna 1771
CAT148 heart 2920 lncrna 1772
CAT148 kich 2920 lncrna 1773
MIR4435-1HG.4 kirc 2901 lncrna 1774
MIR4435-1HG.4 kirp 2901 lncrna 1775
MIR4435-1HG.4 liver 2901 lncrna 1776
MIR4435-1HG.5 kirc 11167 lncrna 1777
MIR4435-1HG.5 stomach 11167 lncrna 1778
CAT294 thyroid 3095 lncrna 1779
CAT294 kirc 3095 lncrna 1780
CAT294 liver 3095 lncrna 1781
MIR4435-1HG.6 head neck 4054 lncrna 1782
MIR4435-1HG.6 stomach 4054 lncrna 1783
MIR4435-1HG.6 kirc 4054 lncrna 1784
MIR4435-1HG.7 kirc 3676 lncrna 1785
MIR4435-1HG.8 kirc 4586 lncrna 1786
OVAT65.3 ovarian 1423 lncrna 1787
CAT969.4 thyroid 984 lncrna 1788
IL10RB-AS1.1 kirc 2299 lncrna 1789
PRCAT119 prostate 10026 lncrna 1790
PRCAT119 prostate 10026 lncrna 1791
CAT972.2 kirc 1161 lncrna 1792
CAT972.2 kirp 1161 lncrna 1793
CAT972.2 kirc 1161 lncrna 1794
CAT972.2 kich 1161 lncrna 1795
CAT972.3 kirc 1282 lncrna 1796
AMAT24 ami 2993 lncrna 1797
KHCAT3.1 kich 975 lncrna 1798
ESAT76.2 embryonic stem cells 12107 lncrna 1799
ESAT76.1 embryonic stem cells 12103 lncrna 1800
CAT 1354 cml 649 lncrna 1801
CAT 1354 kirc 649 lncrna 1802
CAT252.3 medulloblastoma 11801 lncrna 1803
CAT 1966.1 cml 5697 lncrna 1804
CAT 1966.1 kirc 5697 lncrna 1805
SMAT14 skeletal muscle 456 lncrna 1806
LSCAT1.1 lusc 1196 lncrna 1807
CAT1501.3 kirc 656 lncrna 1808
CAT1501.3 kirp 656 lncrna 1809
CAT1501.3 kich 656 lncrna 1810
CAT1501.3 pancreatic 656 lncrna 1811
CAT1501.4 kich 513 lncrna 1812
CAT1501.4 medulloblastoma 513 lncrna 1813
CAT1501.5 medulloblastoma 2898 lncrna 1814
CAT119 colorectal 1750 lncrna 1815
CAT119 uterine 1750 lncrna 1816
LVCAT6 liver 4958 lncrna 1817
CRAT4 colorectal 809 lncrna 1818
MEAT29.2 melanoma 3541 lncrna 1819
MEAT29.1 melanoma 739 lncrna 1820
LINC00948.2 pancreatic 1327 lncrna 1821
LINC00948.2 kirp 1327 lncrna 1822
CAT1410 pancreatic 626 lncrna 1823
CAT1410 colorectal 626 lncrna 1824
PRCAT42.3 prostate 2607 lncrna 1825
PRCAT42.1 prostate 4241 lncrna 1826
OVAT148 ovarian 612 lncrna 1827
CAT1345.3 head neck 885 lncrna 1828
CAT1345.3 luad 885 lncrna 1829
CAT1345.3 lusc 885 lncrna 1830
CAT1345.4 luad 525 lncrna 1831
CAT1345.4 lusc 525 lncrna 1832
CAT1345.5 head neck 2239 lncrna 1833
CAT1345.5 luad 2239 lncrna 1834
CAT1345.5 lusc 2239 lncrna 1835
CAT2040 uterine 1952 lncrna 1836
CAT2040 kich 1952 lncrna 1837
CAT2040 liver 1952 lncrna 1838
CAT1914 gbm 1746 lncrna 1839
CAT1914 igg 1746 lncrna 1840
CAT1914 mpn 1746 lncrna 1841
CAT1914 ami 1746 lncrna 1842
CAT1914 colorectal 1746 lncrna 1843
CAT1914 luad 1746 lncrna 1844
CD27-AS1.1 kirp 1880 lncrna 1845
CD27-AS1.1 prostate 1880 lncrna 1846
CAT1687 thyroid 1043 lncrna 1847
CAT1687 head neck 1043 lncrna 1848
CAT 1687 breast 1043 lncrna 1849
CAT1687 luad 1043 lncrna 1850
CAT1079.3 gbm 583 lncrna 1851
CAT1079.3 583 lncrna 1852
CAT1079.3 kich 583 lncrna 1853
CAT1011 ami 5218 tucp 1854
CAT1011 thyroid 5218 tucp 1855
CAT 1011 breast 5218 tucp 1856
H1FX-AS1.2 colorectal 6093 lncrna 1857
CAT366.4 thyroid 1568 lncrna 1858
CAT366.4 thyroid 1568 lncrna 1859
CAT505.1 prostate 4300 tucp 1860
CAT505.1 breast 4300 tucp 1861
CAT793.3 head neck 10130 tucp 1862
DGCR10.3 kirc 21073 tucp 1863
CAT1855.1 breast 2092 lncrna 1864
CAT1855.2 breast 1987 lncrna 1865
CAT1855.2 breast 1987 lncrna 1866
PRC1-AS1 colorectal 333 lncrna 1867
PRC1-AS1 uterine 333 lncrna 1868
PRC1-AS1 lusc 333 lncrna 1869
PRC1-AS1 breast 333 lncrna 1870
PRC1-AS1 luad 333 lncrna 1871
PRC1-AS1 liver 333 lncrna 1872
LACAT16.1 luad 4814 tucp 1873
CAT2010 melanoma 4289 lncrna 1874
CAT2010 thyroid 4289 lncrna 1875
CAT2010 lusc 4289 lncrna 1876
CAT773.3 embryonic stem cells 527 lncrna 1877
ESAT42.3 embryonic stem cells 2715 lncrna 1878
ESAT42.2 embryonic stem cells 2712 lncrna 1879
CAT1892.2 bladder 5614 lncrna 1880
CAT221 ovarian 949 lncrna 1881
CAT221 lusc 949 lncrna 1882
CAT2275.3 colorectal 1176 lncrna 1883
HRAT4 heart 3659 lncrna 1884
KCCAT41 kirc 616 lncrna 1885
CAT2082.4 head neck 2265 lncrna 1886
OVAT 114.2 ovarian 5819 lncrna 1887
OVAT 114.4 ovarian 3619 lncrna 1888
OVAT114.3 ovarian 8783 lncrna 1889
CAT260.2 cml 22722 lncrna 1890
CAT260.2 ami 22722 lncrna 1891
CAT260.2 kirc 22722 lncrna 1892
CAT 1595 ami 8430 lncrna 1893
CAT1595 kirc 8430 lncrna 1894
MEAT62.1 melanoma 3376 lncrna 1895
MEAT62.3 melanoma 3304 lncrna 1896
ESAT25 embryonic stem cells 842 lncrna 1897
CAT1841.2 kirc 3261 lncrna 1898
CAT1841.3 colorectal 2718 lncrna 1899
CAT1841.3 breast 2718 lncrna 1900
CAT1841.3 pancreatic 2718 lncrna 1901
CAT1841.3 medulloblastoma 2718 lncrna 1902
CAT1841.4 lusc 2433 lncrna 1903
CAT1841.4 head neck 2433 lncrna 1904
CAT1841.4 kirp 2433 lncrna 1905
CAT1841.4 breast 2433 lncrna 1906
CAT1841.4 luad 2433 lncrna 1907
CAT1841.4 kirc 2433 lncrna 1908
CAT1841.4 liver 2433 lncrna 1909
CAT1117 thyroid 1230 lncrna 1910
CAT 1806 kich 1496 lncrna 1911
CAT 1806 thyroid 1496 lncrna 1912
CAT 1806 kirc 1496 lncrna 1913
CAT 1806 kirp 1496 lncrna 1914
CAT1710.1 uterine 1902 tucp 1915
CAT1195.4 kich 8189 lncrna 1916
CAT1195.4 kich 8189 lncrna 1917
CAT1195.4 kirp 8189 lncrna 1918
CAT 1300 mpn 3661 lncrna 1919
CAT 1300 liver 3661 lncrna 1920
CAT 1300 luad 3661 lncrna 1921
CD27-AS1.2 kirp 1758 lncrna 1922
CD27-AS1.2 embryonic stem cells 1758 lncrna 1923
CAT2023 kich 821 lncrna 1924
CAT2023 cervical 821 lncrna 1925
CAT2023 kirc 821 lncrna 1926
CAT2023 kirp 821 lncrna 1927
CAT2023 luad 821 lncrna 1928
CAT2023 lusc 821 lncrna 1929
PNAT23.2 pancreatic 7024 lncrna 1930
PNAT23.5 pancreatic 1409 lncrna 1931
PNAT23.4 pancreatic 13418 tucp 1932
PNAT23.3 pancreatic 13621 tucp 1933
PNAT23.1 pancreatic 1282 lncrna 1934
CAT1701 kich 1055 lncrna 1935
CAT1701 mpn 1055 lncrna 1936
CAT1701 cml 1055 lncrna 1937
CAT1701 ami 1055 lncrna 1938
DGCR5.7 kirc 4192 tucp 1939
DGCR5.7 kirp 4192 tucp 1940
DGCR5.7 luad 4192 tucp 1941
DGCR5.8 kirc 12465 tucp 1942
CAT2051 uterine 1174 lncrna 1943
CAT2051 pancreatic 1174 lncrna 1944
DGCR5.9 kirc 4293 tucp 1945
DGCR5.9 luad 4293 tucp 1946
DGCR5.9 kirc 4293 tucp 1947
CAT742.7 kich 6946 lncrna 1948
CAT742.7 kich 6946 lncrna 1949
CAT1069 ovarian 1412 lncrna 1950
CAT1069 colorectal 1412 lncrna 1951
CAT1069 uterine 1412 lncrna 1952
CAT1069 kirc 1412 lncrna 1953
CAT2068 ovarian 713 lncrna 1954
CAT2068 uterine 713 lncrna 1955
CAT2068 thyroid 713 lncrna 1956
CAT2068 kich 713 lncrna 1957
CAT2068 luad 713 lncrna 1958
CAT2068 breast 713 lncrna 1959
CAT2068 pancreatic 713 lncrna 1960
CAT2012 embryonic stem cells 2410 lncrna 1961
CAT2012 head neck 2410 lncrna 1962
CAT2012 liver 2410 lncrna 1963
CAT2012 luad 2410 lncrna 1964
MP AT 11 mpn 17500 lncrna 1965
CAT1966.2 mpn 5823 lncrna 1966
CAT1966.2 cml 5823 lncrna 1967
CAT1966.2 ami 5823 lncrna 1968
CAT1966.2 kirc 5823 lncrna 1969
CAT1966.2 embryonic stem cells 5823 lncrna 1970
ESAT8.1 embryonic stem cells 4074 lncrna 1971
THCAT50.1 thyroid 1675 lncrna 1972
THCAT50.1 thyroid 1675 lncrna 1973
THCAT50.2 thyroid 1639 lncrna 1974
CAT1224.2 prostate 15239 lncrna 1975
CAT1224.2 breast 15239 lncrna 1976
CAT1224.3 breast 22412 lncrna 1977
CAT120 thyroid 4940 lncrna 1978
CAT120 prostate 4940 lncrna 1979
CAT120 breast 4940 lncrna 1980
CAT120 luad 4940 lncrna 1981
CAT120 lusc 4940 lncrna 1982
CAT120 pancreatic 4940 lncrna 1983
CAT270 heart 423 lncrna 1984
CAT270 embryonic stem cells 423 lncrna 1985
CAT270 head neck 423 lncrna 1986
CAT655.3 head neck 895 lncrna 1987
LACAT3 luad 814 lncrna 1988
ESAT32.2 embryonic stem cells 2299 lncrna 1989
CAT405.1 heart 842 lncrna 1990
CAT405.1 uterine 842 lncrna 1991
CAT405.1 lusc 842 lncrna 1992
HCP5.1 head neck 16291 lncrna 1993
HCP5.1 kich 16291 lncrna 1994
HCP5.1 kirc 16291 lncrna 1995
HCP5.1 skeletal muscle 16291 lncrna 1996
HCP5.2 kirc 13672 lncrna 1997
HCP5.2 skeletal muscle 13672 lncrna 1998
CAT1202.1 ovarian 2314 tucp 1999
CAT1202.2 embryonic stem cells 2066 tucp 2000
CAT 1202.3 ovarian 1079 tucp 2001
CAT1202.3 thyroid 1079 tucp 2002
CAT1202.3 stomach 1079 tucp 2003
CAT1202.3 breast 1079 tucp 2004
ESAT53 embryonic stem cells 3328 lncrna 2005
MIR31HG.4 thyroid 4713 lncrna 2006
CASC .4 head neck 10480 lncrna 2007
CASC9.4 lusc 10480 lncrna 2008
HRAT17.2 heart 818 lncrna 2009
HRAT17.1 heart 867 lncrna 2010
CASC9.5 head neck 1316 lncrna 2011
CASC9.5 luad 1316 lncrna 2012
CASC9.5 lusc 1316 lncrna 2013
HRAT17.5 heart 1579 lncrna 2014
CAT2024 kirc 8337 lncrna 2015
CAT2024 kirc 8337 lncrna 2016
CAT2024 kich 8337 lncrna 2017
UGDH-AS1.4 cervical 8132 tucp 2018
UGDH-AS1.4 thyroid 8132 tucp 2019
CAT505.2 prostate 4033 tucp 2020
CAT505.2 breast 4033 tucp 2021
KDM4A-AS1.2 lusc 5308 lncrna 2022
KDM4A-AS1.3 uterine 3382 lncrna 2023
OVAT150 ovarian 271 lncrna 2024
CAT2168.2 prostate 3715 lncrna 2025
CAT2168.2 breast 3715 lncrna 2026
CAT2168.3 breast 831 lncrna 2027
CAT2168.4 breast 1058 lncrna 2028
CAT2168.5 breast 1098 lncrna 2029
CAT2168.6 breast 976 lncrna 2030
CAT505.3 breast 4286 tucp 20 1
CAT784.1 kich 1094 lncrna 2032
CAT1141.3 thyroid 4764 tucp 2033
CAT1141.4 medulloblastoma 4002 tucp 2034
PRCAT55 prostate 1063 lncrna 2035
PRCAT55 prostate 1063 lncrna 2036 CCAT10 kirc 825 lncrna 2037
KCCAT10 kirc 825 lncrna 2038
CAT1636.2 kirc 484 lncrna 2039
CAT1636.2 liver 484 lncrna 2040
CAT2069.2 thyroid 1070 tucp 2041
CAT2069.2 head neck 1070 tucp 2042
CAT2069.2 kirp 1070 tucp 2043
CAT2069.2 luad 1070 tucp 2044
MIAT.2 embryonic stem cells 4074 lncrna 2045
MIAT.2 head neck 4074 lncrna 2046
MIAT.2 breast 4074 lncrna 2047
MIAT.2 kirc 4074 lncrna 2048
THCAT36.6 thyroid 1180 lncrna 2049
THCAT36.6 thyroid 1180 lncrna 2050
PRCAT102.1 prostate 3656 lncrna 2051
THCAT36.7 thyroid 1170 lncrna 2052
THCAT36.7 thyroid 1170 lncrna 2053
CAT2176.5 head neck 12721 lncrna 2054
LINC00511.5 luad 13108 lncrna 2055
LINC00511.5 lusc 13108 lncrna 2056
LINC00673 luad 7751 lncrna 2057
LINC00673 lusc 7751 lncrna 2058
CALML3-AS1.3 lusc 13476 tucp 2059
CALML3-AS1.4 lusc 9287 tucp 2060
CALML3-AS1.5 lusc 7874 tucp 2061
CALML3-AS1.5 pancreatic 7874 tucp 2062
CALML3-AS1.5 prostate 7874 tucp 2063
CALML3-AS1.6 lusc 9117 tucp 2064
CALML3-AS1.7 lusc 11578 tucp 2065
ESAT73.2 embryonic stem cells 11801 lncrna 2066
ESAT73.3 embryonic stem cells 19092 lncrna 2067
ESAT73.1 embryonic stem cells 11767 lncrna 2068
CAT678 uterine 392 lncrna 2069
CAT678 lusc 392 lncrna 2070
LSCAT8 lusc 894 lncrna 2071
ESAT16.1 embryonic stem cells 3069 lncrna 2072
ESAT16.2 embryonic stem cells 5948 lncrna 2073
ESAT16.3 embryonic stem cells 3 19 lncrna 2074
CAT808 skeletal muscle 3066 lncrna 2075
CAT808 medulloblastoma 3066 lncrna 2076
CAT808 lusc 3066 lncrna 2077
CAT808 kirp 3066 lncrna 2078
CAT808 breast 3066 lncrna 2079
CAT808 luad 3066 lncrna 2080
CAT808 kirc 3066 lncrna 2081
CAT808 kich 3066 lncrna 2082
CAT2045.3 kirc 3770 lncrna 2083
CAT2045.4 rapn 6943 tucp 2084
CAT2045.4 kirc 6943 tucp 2085
CAT1204.5 lusc 1366 lncrna 2086
CAT1204.5 prostate 1366 lncrna 2087
CAT1204.6 thyroid 1329 lncrna 2088
CAT1204.6 prostate 1329 lncrna 2089
ESAT94.2 embryonic stem cells 8025 lncrna 2090
PRCAT89 prostate 2337 lncrna 2091
CAT201.3 kich 1789 lncrna 2092
AMAT7 ami 411 lncrna 2093
CAT1452.2 kich 1906 tucp 2094
OVAT20.2 ovarian 8749 tucp 2095 CCAT63 kirc 1938 tucp 2096 CCAT63 kirc 1938 tucp 2097
CAT955 head neck 4596 lncrna 2098
CAT955 lusc 4596 lncrna 2099
CAT955 luad 4596 lncrna 2100
CAT955 kirc 4596 lncrna 2101
CAT369 luad 2102 lncrna 2102
CAT369 melanoma 2102 lncrna 2103
CAT369 skeletal muscle 2102 lncrna 2104
CAT369 medulloblastoma 2102 lncrna 2105
ESAT89 embryonic stem cells 10862 lncrna 2106
LINC00957 kich 3721 tucp 2107
LINC00957 embryonic stem cells 3721 tucp 2108
HNCAT99 head neck 2534 lncrna 2109
HNCAT3.2 head neck 6556 lncrna 2110
OVAT19 ovarian 341 lncrna 2111
CAT1916 kirc 971 lncrna 2112
CAT1916 head neck 971 lncrna 2113
CAT1916 prostate 971 lncrna 2114
CAT877 mpn 1598 lncrna 2115
CAT877 kirc 1598 lncrna 2116
CAT877 medulloblastoma 1598 lncrna 2117
CAT877 thyroid 1598 lncrna 2118
CAT877 breast 1598 lncrna 2119
CAT877 luad 1598 lncrna 2120
CAT877 lusc 1598 lncrna 2121
CAT1429 kich 932 lncrna 2122
CAT1429 thyroid 932 lncrna 2123
CAT1429 kirc 932 lncrna 2124
CAT1429 kirp 932 lncrna 2125
CAT57.3 thyroid 3492 tucp 2126
CAT57.3 kich 3492 tucp 2127
CAT57.4 thyroid 3093 tucp 2128
TRAF3IP2-AS1.1 gbm 2387 lncrna 2129
TRAF3IP2-AS1.1 igg 2387 lncrna 2130
TRAF3IP2-AS1.1 prostate 2387 lncrna 2131
TRAF3IP2-AS1.1 breast 2387 lncrna 2132
TRAF3IP2-AS1.2 igg 5721 lncrna 2133
TRAF3IP2-AS1.2 breast 5721 lncrna 2134
TRAF3IP2-AS1.3 gbm 5841 lncrna 2135
TRAF3IP2-AS1.3 igg 5841 lncrna 2136
TRAF3IP2-AS1.3 kich 5841 lncrna 2137
CAT781 head neck 8619 lncrna 2138
CAT781 breast 8619 lncrna 2139
CAT781 luad 8619 lncrna 2140
MIR205HG.8 lusc 3923 lncrna 2141
MIR205HG.9 lusc 4139 lncrna 2142
CAT2044 colorectal 356 lncrna 2143
CAT2044 uterine 356 lncrna 2144
AMAT31 ami 1230 lncrna 2145
KPCAT2.2 kirp 1958 lncrna 2146
LINC00511.6 luad 11120 lncrna 2147
SMAT5 skeletal muscle 3550 lncrna 2148
CAT1918 colorectal 808 lncrna 2149
CAT1918 uterine 808 lncrna 2150
CAT1918 kirc 808 lncrna 2151
CAT1918 pancreatic 808 lncrna 2152
ESAT54.2 embryonic stem cells 2476 lncrna 2153
ESAT54.3 embryonic stem cells 6385 lncrna 2154
ESAT54.4 embryonic stem cells 5819 lncrna 2155
ESAT54.6 embryonic stem cells 7677 lncrna 2156
ESAT54.1 embryonic stem cells 10005 lncrna 2157
CAT275 ovarian 644 lncrna 2158
CAT275 breast 644 lncrna 2159
CAT275 cml 644 lncrna 2160
CAT1113.2 lusc 9018 lncrna 2161
CAT1113.3 cervical 6154 lncrna 2162
CAT1113.3 head neck 6154 lncrna 2163
CAT1113.3 luad 6154 lncrna 2164
CAT1113.3 lusc 6154 lncrna 2165
CAT118.2 gbra 1883 tucp 2166
CAT2038 uterine 2445 tucp 2167
CAT2038 thyroid 2445 tucp 2168
DDX11-AS1.3 lusc 920 lncrna 2169
CAT1822 ami 2450 lncrna 2170
CAT1822 kirc 2450 lncrna 2171
CAT708 embryonic stem cells 8558 lncrna 2172
CAT708 stomach 8558 lncrna 2173
ESAT43 embryonic stem cells 764 lncrna 2174
KCCAT101 kirc 3308 lncrna 2175
KCCAT101 kirc 3308 lncrna 2176
SMAT17 skeletal muscle 915 lncrna 2177
LIMD1-AS1.2 thyroid 1896 lncrna 2178
LIMD1-AS1.2 lusc 1896 lncrna 2179
CAT784.2 embryonic stem cells 3134 lncrna 2180
OVAT21 ovarian 1169 tucp 2181
CAT1632 head neck 5529 lncrna 2182
HRAT17.3 heart 531 lncrna 2183
CAT512 igg 12274 tucp 2184
CAT512 kich 12274 tucp 2185
CAT512 kich 12274 tucp 2186
CAT512 kirc 12274 tucp 2187
HRAT17.6 heart 1647 lncrna 2188
KCCAT148.2 kirc 7257 lncrna 2189
KCCAT148.2 kirc 7257 lncrna 2190
KCCAT148.3 kirc 6805 lncrna 2191
PRCAT106 prostate 2039 lncrna 2192
CAT405.2 melanoma 3058 lncrna 2193
CAT62.2 embryonic stem cells 1319 lncrna 2194
CAT62.3 breast 750 lncrna 2195
CAT62.3 luad 750 lncrna 2196
CAT62.3 lusc 750 lncrna 2197
CAT62.4 breast 1508 lncrna 2198
CAT62.4 luad 1508 lncrna 2199
CAT62.4 lusc 1508 lncrna 2200
CAT62.5 embryonic stem cells 1024 lncrna 2201
CAT749.2 kirc 2368 lncrna 2202
CAT749.2 medulloblastoma 2368 lncrna 2203
CAT749.2 stomach 2368 lncrna 2204
CAT749.2 prostate 2368 lncrna 2205
UTAT7 uterine 465 lncrna 2206
MIR205HG.10 lusc 2112 lncrna 2207
MIR205HG.10 breast 2112 lncrna 2208
MIR205HG.i l lusc 1994 lncrna 2209
MIR205HG.i l prostate 1994 lncrna 2210
GATA3-AS1.6 breast 2672 lncrna 2211
GATA3-AS1.6 breast 2672 lncrna 2212
GATA3-AS1.6 kirc 2672 lncrna 2213
ESAT42.4 embryonic stem cells 960 lncrna 2214
TINCR.2 lusc 5928 tucp 2215
ESAT42.1 embryonic stem cells 957 lncrna 2216
CAT1710.2 heart 1269 tucp 2217
CAT1710.2 uterine 1269 tucp 2218
PXN-AS1 embryonic stem cells 707 lncrna 2219
OVAT175 ovarian 993 lncrna 2220
CAT1115.3 lusc 3501 lncrna 2221
CAT1115.4 colorectal 2839 lncrna 2222
CAT1115.4 uterine 2839 lncrna 2223
ESAT54.5 embryonic stem cells 6980 lncrna 2224
ESAT8.2 embryonic stem cells 3913 lncrna 2225
KCCAT192 kirc 15256 lncrna 2226
LVCAT7 liver 1843 lncrna 2227
CAT82 cervical 912 lncrna 2228
CAT82 igg 912 lncrna 2229
CAT82 medulloblastoma 912 lncrna 2230
PRCAT71 prostate 1039 lncrna 2231
PRCAT71 prostate 1039 lncrna 2232
THCAT39.10 thyroid 1098 lncrna 2233
THCAT39.10 thyroid 1098 lncrna 2234
THCAT36.5 thyroid 1421 lncrna 2235
CAT62.6 melanoma 693 lncrna 2236
LINC00511.7 thyroid 3816 lncrna 2237
LINC00511.7 hiad 3816 lncrna 2238
LINC00511.8 luad 4626 lncrna 2239
LINC00511.8 lusc 4626 lncrna 2240
CAT 1193 kirc 3537 lncrna 2241
CAT1193 heart 3537 lncrna 2242
LINC00035 thyroid 745 tucp 2243
LINC000 5 kirp 745 tucp 2244
LINC00035 head neck 745 tucp 2245
LACAT16.2 luad 10365 lncrna 2246
LACAT16.3 luad 6960 lncrna 2247
CAT1508 colorectal 1081 lncrna 2248
CAT1508 uterine 1081 lncrna 2249
CAT1508 breast 1081 lncrna 2250
CAT1508 luad 1081 lncrna 2251
CAT1508 lusc 1081 lncrna 2252
CAT1508 lf¾ 1081 lncrna 2253
CAT76.2 thyroid 2232 lncrna 2254
CAT1383.2 kirc 1109 lncrna 2255
CAT1383.2 breast 1109 lncrna 2256
CAT1383.3 kirc 3577 lncrna 2257
CAT1383.3 breast 3577 lncrna 2258
CAT1383.3 breast 3577 lncrna 2259
CAT147.2 head neck 1660 lncrna 2260
CAT147.2 prostate 1660 lncrna 2261
CAT147.2 breast 1660 lncrna 2262
CAT147.2 luad 1660 lncrna 2263
CAT147.2 medulloblastoma 1660 lncrna 2264
ESAT24.2 embryonic stem cells 2847 lncrna 2265
ESAT24.1 embryonic stem cells 373 lncrna 2266
OVAT114.5 ovarian 6360 lncrna 2267
OVAT114.1 ovarian 4598 lncrna 2268
WT1-AS.8 ovarian 10131 lncrna 2269
WT1-AS.9 ovarian 9999 lncrna 2270
WT1-AS.10 ovarian 10357 lncrna 2271
WT1-AS.11 ovarian 7286 lncrna 2272
WT1-AS.11 kich 7286 lncrna 2273
CAT669 luad 678 lncrna 2274
CAT669 lusc 678 lncrna 2275
ESAT15.1 embryonic_stem_cells 368 lncrna 2276
LBX2-AS1.3 thyroid 2478 tucp 2277
LBX2-AS1.3 head neck 2478 tucp 2278
THCAT36.3 thyroid 3966 lncrna 2279
THCAT36.3 thyroid 3966 lncrna 2280
THCAT36.2 thyroid 1552 lncrna 2281
THCAT36.2 thyroid 1552 lncrna 2282
THCAT36.8 thyroid 1668 lncrna 2283
THCAT36.8 thyroid 1668 lncrna 2284
LINC00511.9 luad 3918 lncrna 2285
LINC00511.9 lusc 3918 lncrna 2286
LINC00511.10 thyroid 3907 lncrna 2287
LINC00511.10 breast 3907 lncrna 2288
LIN C00511.10 luad 3907 lncrna 2289
LINC00511.10 lusc 3907 lncrna 2290
LINC00511.i l lusc 7803 lncrna 2291
CAT249.3 embryonic stem cells 1097 lncrna 2292
BRCAT431.1 breast 1250 lncrna 2293
BRCAT431.1 breast 1250 lncrna 2294
BRCAT431.2 breast 1804 lncrna 2295
BRCAT431.2 breast 1804 lncrna 2296
BRCAT431.3 breast 1333 lncrna 2297
BRCAT431.3 breast 1333 lncrna 2298
BRCAT431.4 breast 2359 lncrna 2299
BRCAT431.4 breast 2359 lncrna 2300
HICLINC62.1 NA 970 lncrna 2301
HICLINC62.2 NA 680 Lncrna 2302
HICLINC62.3 NA 796 lncrna 2303
HICLINC62.4 NA 1273 lncrna 2304
HICLINC62.5 NA 1220 lncrna 2305
HICLINC62.6 NA 1941 lncrna 2306
HICLINC62.7 NA 2041 lncrna 2307
HICLINC62.8 NA 2043 lncrna 2308
HICLINC62.9 NA 2100 lncrna 2309
All publications, patents, patent applications and accession numbers mentioned in the above specification are herein incorporated by reference in their entirety. Although the disclosure has been described in connection with specific embodiments, it should be understood that the disclosure as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications and variations of the described compositions and methods of the disclosure will be apparent to those of ordinary skill in the art and are intended to be within the scope of the following claims.
Claims
We claim: 1. A method of screening for the presence of cancer in a subject, comprising
(a) contacting a biological sample from a subject with a gene expression detection assay, wherein said gene expression detection assay comprises a gene expression informative reagent for identification of the level of expression of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309;
(b) detecting the level of expression of said non-coding in said sample using an in vitro assay; and
(c) diagnosing cancer in said subject when an increased level of expression of said non-coding RNAs in said sample relative to the level in normal cells is detected.
2. The method of claim 1 , wherein the sample is selected from the group consisting of tissue, blood, plasma, serum, urine, urine supernatant, urine cell pellet, semen, prostatic secretions and prostate cells.
3. The method of claim 1 , wherein detection is carried out utilizing a method selected from the group consisting of a sequencing technique, a nucleic acid hybridization technique, and a nucleic acid amplification technique.
4. The method of claim 3, wherein the nucleic acid amplification technique is selected from the group consisting of polymerase chain reaction, reverse transcription polymerase chain reaction, transcription-mediated amplification, ligase chain reaction, strand displacement amplification, and nucleic acid sequence based amplification.
5. The method of claim 1 , wherein said cancer is selected from the group consisting of prostate cancer, breast cancer, acute myeloid leukemia (AML), chronic myeloid leukemia (CML), myeloproliferative neoplasia (MPN)), lower grade glioma (LGG), glioblastome multiforme (GBM)), cervical cancer, head and neck cancer, lung squamous cell cancer, lung adenocarcinoma, kidney cancer, papillary cell carcimona, and bladder cancer.
6. The method of claim 1, wherein said reagent is selected from the group consisiting of a pair of amplification oligonucleotides, a sequencing primer, and an oligonucleotide probe.
7. The method of claim 1, wherein said one or more non-coding RNAs is two or more.
8. The method of claim 1, wherein said one or more non-coding RNAs is ten or more.
9. The method of claim 1 , wherein said one or more non-coding RNAs is 100 or more.
10. The method of claim 1, wherein said non-coding RNAs are converted to cDNA prior to or during detection.
11. The method of claim 6, wherein said reagent comprises one or more labels.
12. A method of identifying gene expression associated with cancer, comprising
(a) contacting a biological sample from a subject with a gene expression detection assay, wherein said gene expression detection assay comprises a gene expression informative reagent for identification of the level of expression of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309;
(b) detecting the level of expression of said non-coding RNA in said sample using an in vitro assay; and
(c) identifying gene expression subjects at risk of prostate cancer metastasis when an increased level of expression of said non-coding RNA said sample relative to the level in normal prostate cells is detected.
13. A system for analyzing a cancer, comprising:
a. a probe set comprising a plurality of probes, wherein the plurality of probes comprises a sequence that hybridizes to at least a portion of one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309 or the corresponding cDNA; and
b. a computer model or algorithm for analyzing an expression level and/or expression profile of said non-coding RNA hybridized to the probe in a sample from a subject.
14. The system of claim 12, further comprising an electronic memory for capturing and storing an expression profile.
15. The system of claim 13, further comprising a computer-processing device, optionally connected to a computer network.
16. The system of claim 13, further comprising a software module executed by the computer- processing device to analyze an expression profile.
17. The system of claim 13, further comprising a software module executed by the computer- processing device to compare the expression profile to a standard or control.
18. The system of claim 13, further comprising a software module executed by the computer- processing device to determine the expression level of the non-coding RNA.
19. The system of claim 13, further comprising a software module executed by the computer- processing device to transmit an analysis of the expression profile to the subject or a medical professional treating the subject.
20. The system of claim 13, further comprising a software module executed by the computer- processing device to transmit a diagnosis or prognosis to the subject or a medical professional treating the subject.
21. The system of claim 13, wherein said one or more non-coding RNAs is two or more.
22. The system of claim 13, wherein said one or more non-coding RNAs is ten or more.
23. The system of claim 13, wherein said one or more non-coding RNAs is 100 or more.
24. The system of claim 13, wherein said probes comprise one or more labels.
25. A probe set for assessing a cancer status of a subject comprising a plurality of probes, wherein the probes in the probe set are capable of detecting an expression level of one or more non- coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309 or the corresponding cDNA.
26. The probe set of claim 25, wherein said plurality of probes comprises five or more probes.
27. The probe set of claim 25, wherein said plurality of probes comprises ten or more probes.
28. The probe set of claim 25, wherein said plurality of probes comprises 100 or more probes.
29. The probe set of claim 25, wherein said probes comprise one or more labels.
30. A composition comprising one or more reaction mixtures, wherein each reaction mixture comprises a complex of a non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309 or the corresponding cDNA and a probe that binds to said non-coding RNA.
31. The composition of claim 30, wherein said one or more reaction mixtures is two or more.
32. The composition of claim 30, wherein said one or more reaction mixtures is ten or more.
33. The composition of claim 30, wherein said one or more reaction mixtures is 100 or more.
34. A method of killing or inhibiting the growth of a cancer cell, comprising contacting a cancer cell with a compound that specifically targets one or more non-coding RNAs selected from the group consisting of those described by SEQ ID NOs: 1-2309.
35. The method of claim 34, wherein said compound is an siRNA or a antisense oligonucleotide.
36. The method of claim 34, wherein said cancer cell is in a subject.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES15867280T ES2807791T3 (en) | 2014-12-08 | 2015-12-08 | Non-coding RNAs and their uses |
| EP15867280.8A EP3230473B1 (en) | 2014-12-08 | 2015-12-08 | Non-coding rnas and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462088772P | 2014-12-08 | 2014-12-08 | |
| US62/088,772 | 2014-12-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016094420A1 true WO2016094420A1 (en) | 2016-06-16 |
Family
ID=56093769
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/064525 WO2016094420A1 (en) | 2014-12-08 | 2015-12-08 | Non-coding rnas and uses thereof |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10889864B2 (en) |
| EP (1) | EP3230473B1 (en) |
| ES (1) | ES2807791T3 (en) |
| WO (1) | WO2016094420A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107029238A (en) * | 2016-11-24 | 2017-08-11 | 汕头大学医学院第附属医院 | Applications of the LINC01094 in diagnosis and treatment cerebral arterial thrombosis |
| CN107058480A (en) * | 2016-12-14 | 2017-08-18 | 河北医科大学第四医院(河北省肿瘤医院) | Long-chain non-coding RNA mark for diagnosing adenocarcinoma of lung |
| CN107130021A (en) * | 2017-05-04 | 2017-09-05 | 南京盖斯夫医药科技有限公司 | The application of CCAT1 long-chains non-coding RNA and its micromolecular inhibitor in terms of hepatocellular carcinoma treatment |
| CN107267636A (en) * | 2017-07-25 | 2017-10-20 | 深圳市龙华区人民医院 | LncRNA GENE NO.9 application and the diagnosticum and healing potion of carcinoma of urinary bladder |
| US10610280B1 (en) | 2019-02-02 | 2020-04-07 | Ayad K. M. Agha | Surgical method and apparatus for destruction and removal of intraperitoneal, visceral, and subcutaneous fat |
| US10815484B2 (en) | 2017-11-22 | 2020-10-27 | The Regents Of The University Of Michigan | Compositions and methods for treating cancer |
| WO2020242080A1 (en) * | 2019-05-28 | 2020-12-03 | 한양대학교 산학협력단 | Squamous cell cancer diagnostic or prognosis prediction marker and use thereof |
| US10889864B2 (en) | 2014-12-08 | 2021-01-12 | The Regents Of The University Of Michigan | Non-coding RNAS and uses thereof |
| US11013754B2 (en) | 2018-04-10 | 2021-05-25 | The Regents Of The University Of Michigan | Compositions and methods for treating cancer |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016081712A1 (en) * | 2014-11-19 | 2016-05-26 | Bigdatabio, Llc | Systems and methods for genomic manipulations and analysis |
| CN107164531A (en) * | 2017-07-05 | 2017-09-15 | 曹巍 | A kind of related serum LncRNA marks of screening lung cancer and its application |
| CN108034655B (en) * | 2017-10-26 | 2021-04-27 | 南京医科大学第二附属医院 | Application of long non-coding RNA and composition thereof in diagnosis/treatment of colorectal cancer |
| EP3539975A1 (en) * | 2018-03-15 | 2019-09-18 | Fundació Privada Institut d'Investigació Oncològica de Vall-Hebron | Micropeptides and uses thereof |
| US20220010310A1 (en) * | 2018-11-09 | 2022-01-13 | The Brigham And Women`S Hospital, Inc. | An lncRNA integrates a DNA-PK-mediated DNA damage response and vascular senescence |
| CN109762821B (en) * | 2019-02-28 | 2021-10-01 | 中山大学孙逸仙纪念医院 | Interfering RNA for inhibiting expression of AFAP1-AS1 and application of interfering RNA in increasing sensitivity of breast cancer radiotherapy |
| CN111826433B (en) * | 2019-04-23 | 2023-08-18 | 清华大学深圳研究生院 | Application of LncRNA in prognosis evaluation of diabetes and early warning of recurrent abortion |
| CN110580934B (en) * | 2019-07-19 | 2022-05-10 | 南方医科大学 | Pregnancy related disease prediction method based on peripheral blood free DNA high-throughput sequencing |
| WO2021095798A1 (en) * | 2019-11-15 | 2021-05-20 | 公立大学法人横浜市立大学 | Sensitive detection method for undifferentiated marker genes |
| CN111254195B (en) * | 2020-01-21 | 2022-11-11 | 上海交通大学医学院附属仁济医院 | Biomarker and kit for predicting chemotherapy effect of colorectal cancer and application of biomarker and kit |
| CN113355411B (en) * | 2020-03-02 | 2022-05-10 | 中山大学孙逸仙纪念医院 | Tumor immunotyping method based on lncRNA marker |
| CN111455056A (en) * | 2020-04-29 | 2020-07-28 | 余之刚 | lncRNA marker derived from adipose cell exosome and application and product thereof |
| CN111944900A (en) * | 2020-08-04 | 2020-11-17 | 佛山科学技术学院 | Characteristic lincRNA expression profile combination and early endometrial cancer prediction method |
| CN115247212B (en) * | 2020-11-06 | 2023-06-06 | 南京市第二医院 | lncRNA for treating, diagnosing and prognosing gastric cancer and application thereof |
| WO2022159383A1 (en) * | 2021-01-19 | 2022-07-28 | President And Fellows Of Harvard College | Engineered nucleic acids targeting long noncoding rna involved in pathogenic infection |
| KR20230165908A (en) * | 2021-03-08 | 2023-12-05 | 후지필름 셀룰러 다이내믹스, 인코포레이티드 | Markers specific to pluripotent stem cells and methods of using the same |
| CN113234815B (en) * | 2021-03-18 | 2022-06-14 | 西北大学 | Application of lncRNA molecule in GBM |
| CN113481296A (en) * | 2021-05-12 | 2021-10-08 | 广东省人民医院 | Diagnostic kit for early evaluation of breast cancer risk |
| CN113736887A (en) * | 2021-09-16 | 2021-12-03 | 南京鼓楼医院集团宿迁医院有限公司 | Application of long-chain non-coding RNA in preparation of esophageal squamous carcinoma precancerous lesion early warning detection reagent |
| CN116622848A (en) * | 2023-05-24 | 2023-08-22 | 广东药科大学 | Application and product of long-chain non-coding RNATUG1 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140073525A1 (en) * | 2011-05-13 | 2014-03-13 | The Board Of Trustees Of The Leland Stanford Junior University | Diagnostic, prognostic and therapeutic uses of long non-coding rnas for cancer and regenerative medicine |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014205555A1 (en) * | 2013-06-28 | 2014-12-31 | British Columbia Cancer Agency Branch | Methods and uses for diagnosis and treatment of prostate cancer |
| US10889864B2 (en) | 2014-12-08 | 2021-01-12 | The Regents Of The University Of Michigan | Non-coding RNAS and uses thereof |
| CA3000775A1 (en) | 2015-07-08 | 2017-01-12 | Dana-Farber Cancer Institute, Inc. | Compositions and methods for identification, assessment prevention, and treatment of cancer using slncr isoforms |
| US20190194150A1 (en) | 2016-07-01 | 2019-06-27 | Arrakis Therapeutics, Inc. | Compounds and methods for modulating rna function |
| AU2018372906A1 (en) | 2017-11-22 | 2020-06-11 | The Regents Of The University Of Michigan | Compositions and methods for treating cancer |
| US11013754B2 (en) | 2018-04-10 | 2021-05-25 | The Regents Of The University Of Michigan | Compositions and methods for treating cancer |
-
2015
- 2015-12-08 US US14/962,961 patent/US10889864B2/en active Active
- 2015-12-08 ES ES15867280T patent/ES2807791T3/en active Active
- 2015-12-08 EP EP15867280.8A patent/EP3230473B1/en active Active
- 2015-12-08 WO PCT/US2015/064525 patent/WO2016094420A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140073525A1 (en) * | 2011-05-13 | 2014-03-13 | The Board Of Trustees Of The Leland Stanford Junior University | Diagnostic, prognostic and therapeutic uses of long non-coding rnas for cancer and regenerative medicine |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE Genbank 13 December 2012 (2012-12-13), "Human DNA sequence from clone RP4-758J18 on chromosome 1p36.31-36.33, complete sequence", XP055454585, Database accession no. AL391244 * |
| See also references of EP3230473A4 * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10889864B2 (en) | 2014-12-08 | 2021-01-12 | The Regents Of The University Of Michigan | Non-coding RNAS and uses thereof |
| CN107029238A (en) * | 2016-11-24 | 2017-08-11 | 汕头大学医学院第附属医院 | Applications of the LINC01094 in diagnosis and treatment cerebral arterial thrombosis |
| CN107029238B (en) * | 2016-11-24 | 2018-08-14 | 汕头大学医学院第一附属医院 | Applications of the LINC01094 in diagnosis and treatment cerebral arterial thrombosis |
| CN107058480A (en) * | 2016-12-14 | 2017-08-18 | 河北医科大学第四医院(河北省肿瘤医院) | Long-chain non-coding RNA mark for diagnosing adenocarcinoma of lung |
| CN107058480B (en) * | 2016-12-14 | 2018-07-13 | 河北医科大学第四医院(河北省肿瘤医院) | Long-chain non-coding RNA marker for diagnosing adenocarcinoma of lung |
| CN107130021A (en) * | 2017-05-04 | 2017-09-05 | 南京盖斯夫医药科技有限公司 | The application of CCAT1 long-chains non-coding RNA and its micromolecular inhibitor in terms of hepatocellular carcinoma treatment |
| CN107130021B (en) * | 2017-05-04 | 2020-06-05 | 中昱医学检验(广州)有限公司 | Application of CCAT1 long-chain non-coding RNA and small-molecule inhibitor thereof in hepatocellular carcinoma treatment |
| CN107267636A (en) * | 2017-07-25 | 2017-10-20 | 深圳市龙华区人民医院 | LncRNA GENE NO.9 application and the diagnosticum and healing potion of carcinoma of urinary bladder |
| CN107267636B (en) * | 2017-07-25 | 2020-05-08 | 深圳市龙华区人民医院 | Application of LncRNA GENE NO.9 and diagnostic agent and therapeutic agent for bladder cancer |
| US10815484B2 (en) | 2017-11-22 | 2020-10-27 | The Regents Of The University Of Michigan | Compositions and methods for treating cancer |
| US11013754B2 (en) | 2018-04-10 | 2021-05-25 | The Regents Of The University Of Michigan | Compositions and methods for treating cancer |
| US10610280B1 (en) | 2019-02-02 | 2020-04-07 | Ayad K. M. Agha | Surgical method and apparatus for destruction and removal of intraperitoneal, visceral, and subcutaneous fat |
| WO2020242080A1 (en) * | 2019-05-28 | 2020-12-03 | 한양대학교 산학협력단 | Squamous cell cancer diagnostic or prognosis prediction marker and use thereof |
| EP3978631A4 (en) * | 2019-05-28 | 2023-06-28 | Industry-University Cooperation Foundation Hanyang University | Squamous cell cancer diagnostic or prognosis prediction marker and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20160160295A1 (en) | 2016-06-09 |
| US10889864B2 (en) | 2021-01-12 |
| EP3230473A1 (en) | 2017-10-18 |
| EP3230473B1 (en) | 2020-04-29 |
| EP3230473A4 (en) | 2018-10-10 |
| ES2807791T3 (en) | 2021-02-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3230473B1 (en) | Non-coding rnas and uses thereof | |
| US20230220485A1 (en) | Cancer biomarkers and classifiers and uses thereof | |
| US20220349011A1 (en) | Cancer diagnostics using biomarkers | |
| US10407735B2 (en) | Schlap-1 ncRNA and uses thereof | |
| EP3571322B1 (en) | Molecular subtyping, prognosis, and treatment of bladder cancer | |
| US11873532B2 (en) | Subtyping prostate cancer to predict response to hormone therapy | |
| US20190100805A1 (en) | Thyroid cancer diagnostics | |
| CN110506127B (en) | Use of genomic tags to predict responsiveness of prostate cancer patients to post-operative radiation therapy | |
| US20220177974A1 (en) | Genetic signatures to predict prostate cancer metastasis and identify tumor aggressiveness | |
| US20160258026A1 (en) | Cancer biomarkers and classifiers and uses thereof | |
| US20190204322A1 (en) | Molecular subtyping, prognosis and treatment of prostate cancer | |
| JP2022512152A (en) | Transcriptome profiling for breast cancer prognosis | |
| CN117203348A (en) | Methods and genomic classifiers for prognosis and identification of breast cancer in subjects who are unlikely to benefit from radiation therapy | |
| WO2017205972A1 (en) | Systems methods and compositions for predicting metastasis in bladder cancer | |
| WO2023220314A1 (en) | Prognosis and treatment of molecular subtypes of prostate cancer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15867280 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015867280 Country of ref document: EP |