There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
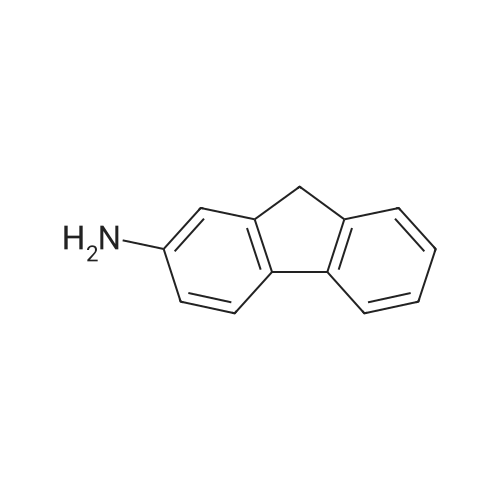
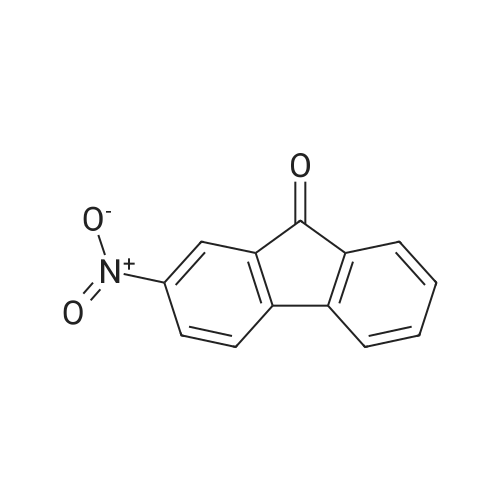
Structure of 607-57-8 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium tetrahydroborate In methanol; water at 0 - 50℃; for 2 h;
General procedure: A mixture of nitroarene (1 mmol), SS-Pd (2 molpercent Pd) and sodium borohydride (3 mmol) were taken in a 25 ml round bottomed flask. 3 ml of MeOH:H2O (3:7) was added to the mixture by a syringe at 0 oC in stirring condition. After 10 minutes the reaction mixture was heated to 50 oC. Progress of the reaction was monitored by TLC. On completion, the reaction mixture was extracted with ethylacetate and dried over anhydrous Na2SO4. Evaporation of the combined organic layer and followed by column chromatography over silica gel (60-120 mesh) afforded desired corresponding amines.
99%
With hydrogen; triethylamine In ethanol; water at 110℃; for 20 h; Autoclave
General procedure: In a 4 ml_ reaction glass vial fitted with a septum cap containing a magnetic stirring bar, Co-Co3O4Chit-700 (10 mg, 3.4 molpercent Co), the nitroarenes (0.5 mmol, 1 .0 equiv.) and triethylamine (35 μΙ_, 0.25 mmol, 0.5 equiv.) were added to a solvent mixture of EtOH/H20 (3/1 , 2 ml_). The reaction vial was then placed into a 300 ml_ autoclave, flashed with hydrogen five times and finally pressurized to 40 bar. The reaction mixture was stirred for appropriate time at 1 10 °C. After cooling the reaction mixture to room temperature, the autoclave was slowly depressurized. The crude reaction mixture was filtered through a pipette fitted with a cotton bed and the solvent was evaporated under reduced pressure. The crude products were purified by passing through a silica plug (eluent: ethyl acetate) to give pure aniline derivatives after removal of solvent. The following compounds may be prepared from the respective nitroarenes using the catalyst of the invention:
97%
With sodium tetrahydroborate In ethanol; water at 25℃; for 4 h;
General procedure: SAC (300mg) and NaBH4 (4.0mmol) were added to a solution of nitroarenes (1.0mmol) in EtOH/water (1/1) (20ml). The reaction mixture was stirred for 4h at the temperature indicated in Table3. At the end of the reaction, the catalyst was removed by filtering and the filtrate was extracted with 3×70ml EtOAc. The combined organic layers were dried over MgSO4 and concentrated in a vacuum.
97%
With iron; ammonium chloride In ethanol; water at 85℃; for 4 h; Reflux; Inert atmosphere
A mixture of 2-nitro-9H-fluorene (2.0 g, 9.5 mmol), iron powder (1.0 g, 18.7 mmol), andNH4Cl (0.75 g, 12.46 mmol) was refluxed in aqueous ethanol(75 mL of alcohol and 25 mL of water) at 85 °C for 4 h under argon atmosphere. The reaction was monitored by TLC (solvent EtOAc–hexane 2:3). After completion of the reaction, theresulting mixture was treated with 50 mL of saturated aqueoussodium bicarbonate solution and filtered off. The transparentfiltrate was concentrated in vacuum in order to remove theorganic solvent. The residue was filtered off to yield compound4(1.67g, 97percent) as transparent plates. The crude product wasused directly in the next step. 1H NMR (CDCl3, 400 MHz) δ7.66 (d, J= 7.6 Hz, 1H), 7.60 (d, J= 8.0 Hz, 1H), 7.50 (d, J =7.2 Hz, 1H), 7.35 (td, J= 7.6 and J= 0.8 Hz, 1H), 7.22 (td, J=7.6 and J= 1.2, 1H), 6.9 (br s, 1H), 6.73 (dd, J= 8.0 and J= 2.4Hz, 1H), 3.84 (s, 2H), 3.70 (br s, 2H, NH2) ppm; 13C NMR(CDCl3, 100 MHz) δ145.75, 145.16, 142.27, 142.15, 133.01,126.64, 125.09, 124.76, 120.67, 118.6, 113.98, 111.82, 36.83ppm.
96%
With sodium tetrahydroborate In ethanol; water at 20℃; for 1.5 h;
General procedure: TAPEHA-Pd (0.015 g) was added to a solution of nitroarenes (1.0 mmol) in EtOH/water (1/1) (20 mL). After NaBH4 (4.0 mmol) was slowly added to the mixture, the color of the reaction mixture turned gradually black in a few minutes, resulting in the formation of palladium nanoparticles (TAPEHA-PdNPs). 42 After being stirredfor 1.5 h at room temperature and atmospheric pressure, the catalyst was removed by ltering and the fitrate was extracted with 3 30 mL of EtOAc. The combined organic layers were dried over MgSO4 and concentrated in a vacuum.
93%
at 80℃; for 0.0833333 h; Microwave irradiation
A mixture of 1j (100 mg, 0.81 mmol), hydrazine hydrate (121.5mg, 2.43 mmol), and SS-Rh (370 mg, 2 molpercent Rh) were taken in an oven dried reaction tube equipped with screw cap. 0.5 ml of PEG-400 was added into the reaction mixture. The reaction was then irradiated in a microwave apparatus at 80°C , 80 W for 10 min with a pressure of 80 Psi. After cooling to ambient temperature in the microwave cavity the reaction mixture was extracted with ethyl acetate (3x2 ml) and water (1ml). The combined organic layer wasdried over anhydrous Na2SO4 and the solvent was removed under reduced pressure and after purificationwith silica gel column chromatography (Hexane: EtOAc::95:5) 2j as brown powder (80 mg, 93percent), m.p.124-125°C. 1H and 13C NMR spectra has beencompared with our previously reported study.3 ESI-MS: m/z calc. for (M+H)+ C13H11N182.2405 and obsd.182.0631
78%
With calcium chloride; zinc In ethanol
Reduction of 2-nitrofluorene with Zn/CaCl2 in ethanol gave 2-aminofluorene (17) in 78percent yield.
62%
With 5%-palladium/activated carbon; hydrazine hydrate In ethanol at 0.5℃; for 3.5 h; Reflux
In a two-necked round-bottomed flask (500 mL) equipped with a reflux condenser and a dropping funnel, a suspension of 2-nitro-9H-fluorene (10.12 g, 48 mmol), palladium on carbon 5percent (5 g), and ethanol (250 mL) was prepared. The mixture was heated, and while being stirred magnetically, hydrazine hydrate 85percent (35 mL) in ethanol (50 mL) was added dropwise over a 1.5 h period through the dropping funnel while maintaining the temperature at about 50 °C. The reaction mixture was then refluxed for 2 h and filtered while hot. On cooling, the filtrate gave light yellow colored crystals of the title diamine compound, which was recrystallized from ethanol and dried under vacuum to give 5.4 g (62percent yield) (Scheme1). mp 129-134 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.66 (1H, d, J = 7.6 Hz), 7.59 (1H, d, J = 8 Hz), 7.49 (1H, d, J = 7.6 Hz), 7.36-7.32 (1H, m), 7.22 (1H, ddd, J = 1.2 Hz, J = 7.6 Hz), 6.91 (1H, t, J = 0.4 Hz), 6.74 (1H, dd, J = 2 Hz, J = 8 Hz), 3.83 (2H, S), 3.76 (2H, S, NH2). 13C NMR (100 MHz, CDCl3), δ, ppm: 145.74, 145.16, 142.26, 142.14, 133, 126.63, 125.08, 124.75, 120.66, 118.58, 113.97, and 111.81. The mass spectrum show peak at m/z = 181.1 corresponding to compound F2 (Figs. S3-S4).
92 %Chromat.
With carbon monoxide; water In tetrahydrofuran at 125℃; for 24 h; Inert atmosphere; Autoclave
General procedure: Into a reaction glass vial fitted with a magnetic stirring bar anda septum cap penetrated with a syringe needle was added theCo3O4/NGrC-catalyst (2 molpercent, 3 wtpercent Co-phenanthroline oncarbon, 20 mg) followed by the nitro arene (0.5 mmol), theinternal standard (hexadecane, 100 μL), THF (2 mL), and H2O(200 μL). The reaction vial was then placed into a 300 mL autoclave.The autoclave was flushed twice with nitrogen, pressurized with CO at 30 bar pressure. Finally, the autoclave was usedat 60 bar by adding nitrogen and placed into an aluminiumblock, which was preheated at 125 °C. After 24 h the autoclavewas placed into a water bath and cooled to r.t. Finally, theremaining gas was discharged, and the samples were removedfrom the autoclave, diluted with EtOAc and analyzed by GC. Todetermine the yield of isolated products, the general procedurewas scaled up by the factor of two, and no internal standard wasadded. After the reaction was completed, the catalyst was filteredoff, and the filtrate was concentrated and purified by silicagel column chromatography (n-heptane–EtOAc mixtures) togive the corresponding anilines.
99 %Chromat.
With 4-amino-phenol; triethylamine In ethanol; water at 110℃; for 9 h; Autoclave
General procedure: In an 8mL glass vial fitted with a magnetic stirring bar and a septum cap, the catalyst (the amount depends on the catalyst) was added followed by the nitroarene (0.5mmol), the internal standard (hexadecane, 20mg) and the solvent (2mL). A needle was inserted in the septum cap, which allows dihydrogen to enter. The vials (up to 7) were placed into a 300mL steel Parr autoclave which was flushed twice with dihydrogen at 20bar and then pressurized to 50bar. Then the autoclave was placed into an aluminum block pre-heated at 110°C. At the end of the reaction, the autoclave was quickly cooled down at room temperature with an ice bath and vented. Finally, the samples were removed from the autoclave, diluted with a suitable solvent, filtered using a Pasteur pipette filled with Celite® (6cm pad) and analyzed by GC using n-hexadecane as an internal standard. Control experiments showed that the position of the vial inside the autoclave is not influential. The same outcome was obtained when the reaction was repeated by moving a vial from a peripheral to a central position.
Reference:
[1] Analytical Chemistry, 1988, vol. 60, # 3, p. 194 - 199
[2] Tetrahedron Letters, 2012, vol. 53, # 36, p. 4858 - 4861
[3] Chemical Communications, 2013, vol. 49, # 86, p. 10088 - 10090
[4] ChemSusChem, 2017, vol. 10, # 15, p. 3035 - 3039
[5] Applied Organometallic Chemistry, 2018, vol. 32, # 1,
[6] Patent: WO2018/114777, 2018, A1, . Location in patent: Page/Page column 31-32
[7] ChemSusChem, 2018, vol. 11, # 18, p. 3131 - 3138
[8] Catalysis Communications, 2015, vol. 67, p. 64 - 67
[9] Beilstein Journal of Organic Chemistry, 2016, vol. 12, p. 1749 - 1757
[10] Advanced Synthesis and Catalysis, 2012, vol. 354, # 2-3, p. 321 - 327
[11] Turkish Journal of Chemistry, 2017, vol. 41, # 5, p. 784 - 792
[12] Tetrahedron Letters, 1980, vol. 21, # 27, p. 2603 - 2604
[13] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1988, vol. 24, p. 87 - 92[14] Khimiya Geterotsiklicheskikh Soedinenii, 1988, vol. 24, # 1, p. 104 - 109
[15] Tetrahedron Letters, 2014, vol. 55, # 18, p. 2912 - 2916
[16] Tetrahedron Letters, 2000, vol. 41, # 30, p. 5603 - 5606
[17] ChemCatChem, 2017, vol. 9, # 6, p. 1128 - 1134
[18] Synthetic Communications, 2000, vol. 30, # 20, p. 3745 - 3754
[19] Heterocycles, 2004, vol. 63, # 2, p. 283 - 296
[20] Patent: US6362200, 2002, B1, . Location in patent: Example 2
[21] Catalysis Letters, 2014, vol. 144, # 7, p. 1258 - 1267
[22] European Journal of Organic Chemistry, 2016, vol. 2016, # 20, p. 3307 - 3309
[23] Bulletin of the Korean Chemical Society, 2010, vol. 31, # 4, p. 984 - 988
[24] Patent: US2011/77394, 2011, A1, . Location in patent: Page/Page column 15
[25] Chemistry - A European Journal, 2016, vol. 22, # 32, p. 11291 - 11302
[26] Journal of Organometallic Chemistry, 1981, vol. 219, # 1, p. 125 - 127
[27] European Journal of Organic Chemistry, 2011, # 4, p. 672 - 675
[28] Journal of Nanoscience and Nanotechnology, 2018, vol. 18, # 11, p. 7873 - 7881
[29] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2018, vol. 189, p. 22 - 31
[30] New Journal of Chemistry, 2018, vol. 42, # 2, p. 1373 - 1378
[31] Chemical and Pharmaceutical Bulletin, 1983, vol. 31, # 2, p. 776 - 779
[32] Chemische Berichte, 1901, vol. 34, p. 1759
[33] Organic Syntheses, 1960, vol. 40, p. 5
[34] Journal of the American Chemical Society, 1947, vol. 69, p. 712
[35] Journal of the American Chemical Society, 1949, vol. 71, p. 1893 Anm.2
[36] Canadian Journal of Chemistry, 1957, vol. 35, p. 1084,1085
[37] Zhurnal Obshchei Khimii, 1957, vol. 27, p. 234,237;engl.Ausg.S.261,264
[38] Journal of Organic Chemistry, 1958, vol. 23, p. 680,683
[39] Journal of the Chemical Society, 1957, p. 207
[40] Journal of the Chemical Society, 1955, p. 2686,2688
[41] Chemische Berichte, 1884, vol. 17, p. 107
[42] Journal of the Chemical Society, 1908, vol. 93, p. 1763
[43] Organic Syntheses, 1933, vol. 13, p. 75[44] Coll.Vol., 1943, vol. II, p. 448
[45] Canadian Journal of Chemistry, 1957, vol. 35, p. 1084,1085
[46] Zhurnal Obshchei Khimii, 1957, vol. 27, p. 234,237;engl.Ausg.S.261,264
[47] Journal of Organic Chemistry, 1958, vol. 23, p. 680,683
[48] Molecular Crystals and Liquid Crystals (1969-1991), 1986, vol. 133, p. 111 - 124
[49] Chemical Research in Toxicology, 2000, vol. 13, # 8, p. 793 - 800
[50] Chemical Research in Toxicology, 1998, vol. 11, # 11, p. 1361 - 1367
[51] Chemical Research in Toxicology, 2002, vol. 15, # 4, p. 536 - 544
[52] Bulletin de la Societe Chimique de France, 1927, vol. <4> 41, p. 69
[53] Chemical Communications, 2010, vol. 46, # 10, p. 1769 - 1771
[54] Advanced Synthesis and Catalysis, 2011, vol. 353, # 8, p. 1306 - 1316
[55] Chemistry - A European Journal, 2011, vol. 17, # 21, p. 5903 - 5907
[56] Chemical Communications, 2011, vol. 47, # 39, p. 10972 - 10974
[57] Chemical Communications, 2013, vol. 49, # 80, p. 9089 - 9091
[58] Synlett, 2015, vol. 26, # 3, p. 313 - 317
[59] ACS Catalysis, 2015, vol. 5, # 3, p. 1526 - 1529
[60] Chemical Communications, 2015, vol. 51, # 66, p. 13086 - 13089
[61] RSC Advances, 2015, vol. 5, # 105, p. 86529 - 86535
[62] Catalysis Science and Technology, 2016, vol. 6, # 12, p. 4473 - 4477
[63] Journal of Catalysis, 2017, vol. 351, p. 79 - 89
[64] Applied Catalysis A: General, 2018, vol. 559, p. 127 - 137
[65] Chemistry - A European Journal, 2018, vol. 24, # 70, p. 18682 - 18688
2
[ 607-57-8 ]
[ 24237-69-2 ]
[ 153-78-6 ]
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1980, # 17, p. 821
3
[ 607-57-8 ]
[ 153-78-6 ]
[ 53-96-3 ]
Reference:
[1] Chemical Research in Toxicology, 2002, vol. 15, # 4, p. 536 - 544
4
[ 607-57-8 ]
[ 5405-53-8 ]
Reference:
[1] Journal of Organic Chemistry, 2004, vol. 69, # 3, p. 987 - 990
5
[ 607-57-8 ]
[ 5405-53-8 ]
[ 15110-74-4 ]
Reference:
[1] Annales de Chimie (Cachan, France), 1930, vol. <10> 14, p. 52,55, 95
[2] Journal of the Chemical Society, 1926, p. 2694
[3] Annales de Chimie (Cachan, France), 1930, vol. <10> 14, p. 52,55, 95
[4] Journal of the Chemical Society, 1926, p. 2694
6
[ 607-57-8 ]
[ 7697-37-2 ]
[ 64-19-7 ]
[ 5405-53-8 ]
[ 15110-74-4 ]
Reference:
[1] Annales de Chimie (Cachan, France), 1930, vol. <10> 14, p. 52,55, 95
[2] Journal of the Chemical Society, 1926, p. 2694
With nitric acid; acetic acid at 60℃; for 0.166667h;
2-Nitro-9H-fluorene (3)
9H-Fluorene (6.0 g, 36.1 mmol)was dissolved in 100 mL of glacial acetic acid at 60 °C. 15 mLof nitric acid (65%) were added dropwise (~10 min) at 60 °Cupon vigorous stirring. After the addition was completed, theresulting mixture was further stirred at 60 °C. The reaction wasmonitored by TLC (solvent EtOAc-n-heptane 1:9). After appearance of the spot of the dinitro product (≈100 min, Rf≈25%), the mixture was poured into 600 mL of water. The resulting crude product was filtered off, washed with water andrecrystallized from 200 mL acetonitrile to give compound 3(7.1 g, 92%) as slightly-yellow needles. 1H NMR (CDCl3,400 MHz) δ8.42 (s, 1H), 8.31 (d, J= 8.4 Hz, 1H), 7.89-7.87(m, 2H), 7.64 (d, J= 6.4 Hz, 1H), 7.49-7.43 (m, 2H), 4.02 (s,2H) ppm; 13C NMR (CDCl3, 100 MHz) δ148.09, 146.79,144.81, 143.92, 139.47, 128.87, 127.43, 125.43, 123.14, 121.34,120.49, 119.88, 36.96 ppm.
91%
With nitric acid; acetic acid at 60 - 80℃;
2.3.1 The preparation of 2-nitro-9H-fluorene (1)
The 2-nitro-9H-fluorene (1) was prepared based on the procedure reported in the literature [46]. 9H-fluorene (4.9 g, 30 mmol) was dissolved in 45 mL of glacial acetic acid at 60 °C and subsequently, nitric acid 65 % (7 mL) was added in a dropwise manner over 15 min with vigorous stirring. During the addition, the color of the solution slightly turns yellow, along with a little precipitation. The resulting mixture was allowed to stir at 80 °C. The progress of the reaction was screened via TLC (ethyl acetate: hexane 1:9 v/v, Rf value = 0.67). After completion of the reaction, the mixture was poured into 300 mL of water and then, the crude product was filtered off, washed with water and recrystallized from 150 mL ethanol to afford the pure product 1 (5.78 g, 91 %) as slightly-yellow needles. M.p. 155-157 °C; 1H NMR (CDCl3, 400 MHz) δ 8.43 (s, 1 H), 8.33 (d, J = 8 Hz, 1 H), 7.91-7.88 (m, 2 H), 7.65 (d, J = 6.4 Hz, 1 H), 7.50-7.46 (m, 2 H), 4.04 (s, 2 H) ppm (Fig. S1).
90%
With nitric acid; acetic acid at 50 - 80℃;
90%
With bismuth(III) nitrate for 0.0333333h; Microwave irradiation; regioselective reaction;
Synthesis
General procedure: The synthesis of nitro compounds by bismuth nitrate under various solid-phase conditions using microwave irradiation is shown in Scheme-I. The compound to be nitrated (1 mmol) and solid support were added to a suspension of bismuth nitrate (1 mmol) in THF (5 mL). The solvent was evaporated under reduced pressure and irradiated in a microwave oven for 1 to 2 min. The mixture was then washed with dichloromethane (10 mL) and it was concentrated to afford the crude product. The pure product was isolated after column chromatography. Several aromatic compounds (1), as well a ssolid supports, were considered in this study, and the corresponding products (2) were obtained in good yield.
86%
With bismuth(III) nitrate; Montmorillonite KSF for 0.133333h;
80%
With nitric acid In acetic acid at 85℃;
77%
With nitric acid; acetic acid at 50 - 85℃; for 0.416667h; Inert atmosphere;
2.2.1 2.2.1. Synthesis of 2-Nitro-9H-Fluorene (F1)
Fluorene (60 g, 361 mmol) and 500 mL AcOH were added to a 1 L three-necked flask equipped with a thermometer and addition funnel. The reaction mixture was heated to 50 °C under nitrogen atmosphere. Then, nitric acid (80 mL) was added over 20 min via the addition funnel. The temperature was subsequently raised to 85 °C and maintained for 5 min. The resulting mixture was removed from the heater and then permitted to cool to room temperature over 2 h. Then, the resulting yellow suspension was filtered, washed with 50 mL AcOH containing 1.3 g KOAc, slurried in water and filtered again. The yellow product was dried in a vacuum oven, affording 58 g (77% yields) of desired product. The melting point was 152-158 °C. (Lit 155-156 °C) [47] . 1H NMR (400 MHz, CDCl3), δ, ppm: 8.42 (1H, d, J = 1.2 Hz), 8.31 (1H, dd, J = 2 Hz, J = 8 Hz), 7.90-7.87 (2H, m), 7.65-7.62 (1H, m), 7.49-7.43 (2H, m), 4.01 (2H, S). 13C NMR (100 MHz, CDCl3), δ, ppm: 148.08, 146.74, 144.82, 143.92, 139.46, 128.87, 127.43, 125.43, 123.13, 121.34, 120.48, and 119.88. The mass spectrum show peak at m/z = 211.1 corresponding to compound F1 (Figs. S1-S2).
74%
With thionyl chloride; bismuth subnitrate In dichloromethane at 20℃; for 2h;
73%
With nitric acid; acetic acid at 20 - 80℃; for 2.33h;
46%
With sulfuric acid; nitric acid; silica gel In dichloromethane at 20℃; for 96h;
42%
With nitric acid; acetic acid
15%
With nitric acid In acetic acid at -43℃; for 6h;
With dinitrogen trioxide; Petroleum ether at 0 - 30℃;
With dinitrogen trioxide; benzene at 0 - 30℃;
With dinitrogen tetraoxide; Petroleum ether
With nitric acid at 20 - 25℃;
With nitric acid; acetic acid
With nitric acid; acetic acid
With nitric acid; acetic acid at 50℃; anschliessend Erwaermen auf 80-85grad;
(nitration);
10 g
With nitric acid; acetic acid at 60 - 80℃;
11 g
With nitric acid In 1,2-dichloro-ethane for 0.333333h; Cooling with ice;
1
Add 250 mL to a 250 mL flask1,2-dichloroethane, and then 20 mL of concentrated nitric acid (65%) was added to disperse all The reaction was carried out in an ice-water bath followed by the slow addition of 10 g of fluorene. After 20 min reaction, the mixture was poured into 300 mL of ice methanol and filtered to give pale yellow solid 2 (11 g).A solution of 7.5 g of KOH was added to 100 mL of DMSO and dispersed uniformly After the addition of the above pale yellow solid 6.3 g, stirring for 20 min, 15 mL of 1-bromooctane was added dropwise to the above system, The reaction was carried out for 30 min, extracted with petroleum ether, dried and dried to give the crude product, which was then subjected to column chromatography using petroleum ether as developing solvent A yellow viscous liquid, compound A (9.0 g) was obtained.
60 g
With nitric acid In acetic acid at 50 - 70℃;
1 Example 1 : Preparation of compound 1
0184] A mixture of fluorene (60 g) in AcOH (500 mL) was mechanically stirred in a 3-neck round-bottom flask and the temperature of the solution was raised to an internal temperature of 50 °C. Then 70% nitric acid (aq) was added dropwise over the course of 15 minutes with continuous stirring. The temperature was raised to 70°C and solid product began to form from the clear mixture. The reaction was monitored by TLC until a lower Rf yellow spot formed (silica, 4/6, chloroform/hexane on silica). Once the reaction was complete and no fluorene remained, the reaction was cooled to room temperature. The solid was fdtered out and washed with water (3 x 200 mL). The solid was dried under vacuum. The solid was recrystallized from EtOAc to give pure compound 1 (60 g).
60 g
With nitric acid In acetic acid at 50 - 70℃;
1 Example 1 : Preparation of compound 1
0184] A mixture of fluorene (60 g) in AcOH (500 mL) was mechanically stirred in a 3-neck round-bottom flask and the temperature of the solution was raised to an internal temperature of 50 °C. Then 70% nitric acid (aq) was added dropwise over the course of 15 minutes with continuous stirring. The temperature was raised to 70°C and solid product began to form from the clear mixture. The reaction was monitored by TLC until a lower Rf yellow spot formed (silica, 4/6, chloroform/hexane on silica). Once the reaction was complete and no fluorene remained, the reaction was cooled to room temperature. The solid was fdtered out and washed with water (3 x 200 mL). The solid was dried under vacuum. The solid was recrystallized from EtOAc to give pure compound 1 (60 g).
With sodium tetrahydroborate; In methanol; water; at 0 - 50℃; for 2h;
General procedure: A mixture of nitroarene (1 mmol), SS-Pd (2 mol% Pd) and sodium borohydride (3 mmol) were taken in a 25 ml round bottomed flask. 3 ml of MeOH:H2O (3:7) was added to the mixture by a syringe at 0 oC in stirring condition. After 10 minutes the reaction mixture was heated to 50 oC. Progress of the reaction was monitored by TLC. On completion, the reaction mixture was extracted with ethylacetate and dried over anhydrous Na2SO4. Evaporation of the combined organic layer and followed by column chromatography over silica gel (60-120 mesh) afforded desired corresponding amines.
99%
With hydrogen; triethylamine; In ethanol; water; at 110℃; under 30003 Torr; for 20h;Autoclave;
General procedure: In a 4 ml_ reaction glass vial fitted with a septum cap containing a magnetic stirring bar, Co-Co3O4Chit-700 (10 mg, 3.4 mol% Co), the nitroarenes (0.5 mmol, 1 .0 equiv.) and triethylamine (35 muIota_, 0.25 mmol, 0.5 equiv.) were added to a solvent mixture of EtOH/H20 (3/1 , 2 ml_). The reaction vial was then placed into a 300 ml_ autoclave, flashed with hydrogen five times and finally pressurized to 40 bar. The reaction mixture was stirred for appropriate time at 1 10 C. After cooling the reaction mixture to room temperature, the autoclave was slowly depressurized. The crude reaction mixture was filtered through a pipette fitted with a cotton bed and the solvent was evaporated under reduced pressure. The crude products were purified by passing through a silica plug (eluent: ethyl acetate) to give pure aniline derivatives after removal of solvent. The following compounds may be prepared from the respective nitroarenes using the catalyst of the invention:
97%
With sodium tetrahydroborate; In ethanol; water; at 25℃; for 4h;
General procedure: SAC (300mg) and NaBH4 (4.0mmol) were added to a solution of nitroarenes (1.0mmol) in EtOH/water (1/1) (20ml). The reaction mixture was stirred for 4h at the temperature indicated in Table3. At the end of the reaction, the catalyst was removed by filtering and the filtrate was extracted with 3×70ml EtOAc. The combined organic layers were dried over MgSO4 and concentrated in a vacuum.
97%
With iron; ammonium chloride; In ethanol; water; at 85℃; for 4h;Reflux; Inert atmosphere;
A mixture of 2-nitro-9H-fluorene (2.0 g, 9.5 mmol), iron powder (1.0 g, 18.7 mmol), andNH4Cl (0.75 g, 12.46 mmol) was refluxed in aqueous ethanol(75 mL of alcohol and 25 mL of water) at 85 C for 4 h under argon atmosphere. The reaction was monitored by TLC (solvent EtOAc-hexane 2:3). After completion of the reaction, theresulting mixture was treated with 50 mL of saturated aqueoussodium bicarbonate solution and filtered off. The transparentfiltrate was concentrated in vacuum in order to remove theorganic solvent. The residue was filtered off to yield compound4(1.67g, 97%) as transparent plates. The crude product wasused directly in the next step. 1H NMR (CDCl3, 400 MHz) delta7.66 (d, J= 7.6 Hz, 1H), 7.60 (d, J= 8.0 Hz, 1H), 7.50 (d, J =7.2 Hz, 1H), 7.35 (td, J= 7.6 and J= 0.8 Hz, 1H), 7.22 (td, J=7.6 and J= 1.2, 1H), 6.9 (br s, 1H), 6.73 (dd, J= 8.0 and J= 2.4Hz, 1H), 3.84 (s, 2H), 3.70 (br s, 2H, NH2) ppm; 13C NMR(CDCl3, 100 MHz) delta145.75, 145.16, 142.27, 142.15, 133.01,126.64, 125.09, 124.76, 120.67, 118.6, 113.98, 111.82, 36.83ppm.
96%
With sodium tetrahydroborate; In ethanol; water; at 20℃; under 760.051 Torr; for 1.5h;
General procedure: TAPEHA-Pd (0.015 g) was added to a solution of nitroarenes (1.0 mmol) in EtOH/water (1/1) (20 mL). After NaBH4 (4.0 mmol) was slowly added to the mixture, the color of the reaction mixture turned gradually black in a few minutes, resulting in the formation of palladium nanoparticles (TAPEHA-PdNPs). 42 After being stirredfor 1.5 h at room temperature and atmospheric pressure, the catalyst was removed by ltering and the fitrate was extracted with 3 30 mL of EtOAc. The combined organic layers were dried over MgSO4 and concentrated in a vacuum.
93%
With hydrazine hydrate; at 80℃; under 4137.29 Torr; for 0.0833333h;Microwave irradiation;
A mixture of 1j (100 mg, 0.81 mmol), hydrazine hydrate (121.5mg, 2.43 mmol), and SS-Rh (370 mg, 2 mol% Rh) were taken in an oven dried reaction tube equipped with screw cap. 0.5 ml of PEG-400 was added into the reaction mixture. The reaction was then irradiated in a microwave apparatus at 80C , 80 W for 10 min with a pressure of 80 Psi. After cooling to ambient temperature in the microwave cavity the reaction mixture was extracted with ethyl acetate (3x2 ml) and water (1ml). The combined organic layer wasdried over anhydrous Na2SO4 and the solvent was removed under reduced pressure and after purificationwith silica gel column chromatography (Hexane: EtOAc::95:5) 2j as brown powder (80 mg, 93%), m.p.124-125C. 1H and 13C NMR spectra has beencompared with our previously reported study.3 ESI-MS: m/z calc. for (M+H)+ C13H11N182.2405 and obsd.182.0631
78%
With calcium chloride; zinc; In ethanol;
Reduction of 2-nitrofluorene with Zn/CaCl2 in ethanol gave 2-aminofluorene (17) in 78% yield.
62%
With 5%-palladium/activated carbon; hydrazine hydrate; In ethanol; at 0.5℃; for 3.5h;Reflux;
In a two-necked round-bottomed flask (500 mL) equipped with a reflux condenser and a dropping funnel, a suspension of 2-nitro-9H-fluorene (10.12 g, 48 mmol), palladium on carbon 5% (5 g), and ethanol (250 mL) was prepared. The mixture was heated, and while being stirred magnetically, hydrazine hydrate 85% (35 mL) in ethanol (50 mL) was added dropwise over a 1.5 h period through the dropping funnel while maintaining the temperature at about 50 C. The reaction mixture was then refluxed for 2 h and filtered while hot. On cooling, the filtrate gave light yellow colored crystals of the title diamine compound, which was recrystallized from ethanol and dried under vacuum to give 5.4 g (62% yield) (Scheme1). mp 129-134 C. 1H NMR (400 MHz, CDCl3), delta, ppm: 7.66 (1H, d, J = 7.6 Hz), 7.59 (1H, d, J = 8 Hz), 7.49 (1H, d, J = 7.6 Hz), 7.36-7.32 (1H, m), 7.22 (1H, ddd, J = 1.2 Hz, J = 7.6 Hz), 6.91 (1H, t, J = 0.4 Hz), 6.74 (1H, dd, J = 2 Hz, J = 8 Hz), 3.83 (2H, S), 3.76 (2H, S, NH2). 13C NMR (100 MHz, CDCl3), delta, ppm: 145.74, 145.16, 142.26, 142.14, 133, 126.63, 125.08, 124.75, 120.66, 118.58, 113.97, and 111.81. The mass spectrum show peak at m/z = 181.1 corresponding to compound F2 (Figs. S3-S4).
92%Chromat.
With carbon monoxide; water; In tetrahydrofuran; at 125℃; under 22502.3 - 45004.5 Torr; for 24h;Inert atmosphere; Autoclave;
General procedure: Into a reaction glass vial fitted with a magnetic stirring bar anda septum cap penetrated with a syringe needle was added theCo3O4/NGrC-catalyst (2 mol%, 3 wt% Co-phenanthroline oncarbon, 20 mg) followed by the nitro arene (0.5 mmol), theinternal standard (hexadecane, 100 muL), THF (2 mL), and H2O(200 muL). The reaction vial was then placed into a 300 mL autoclave.The autoclave was flushed twice with nitrogen, pressurized with CO at 30 bar pressure. Finally, the autoclave was usedat 60 bar by adding nitrogen and placed into an aluminiumblock, which was preheated at 125 C. After 24 h the autoclavewas placed into a water bath and cooled to r.t. Finally, theremaining gas was discharged, and the samples were removedfrom the autoclave, diluted with EtOAc and analyzed by GC. Todetermine the yield of isolated products, the general procedurewas scaled up by the factor of two, and no internal standard wasadded. After the reaction was completed, the catalyst was filteredoff, and the filtrate was concentrated and purified by silicagel column chromatography (n-heptane-EtOAc mixtures) togive the corresponding anilines.
99%Chromat.
With 4-amino-phenol; triethylamine; In ethanol; water; at 110℃; under 37503.8 Torr; for 9h;Autoclave;
General procedure: In an 8mL glass vial fitted with a magnetic stirring bar and a septum cap, the catalyst (the amount depends on the catalyst) was added followed by the nitroarene (0.5mmol), the internal standard (hexadecane, 20mg) and the solvent (2mL). A needle was inserted in the septum cap, which allows dihydrogen to enter. The vials (up to 7) were placed into a 300mL steel Parr autoclave which was flushed twice with dihydrogen at 20bar and then pressurized to 50bar. Then the autoclave was placed into an aluminum block pre-heated at 110C. At the end of the reaction, the autoclave was quickly cooled down at room temperature with an ice bath and vented. Finally, the samples were removed from the autoclave, diluted with a suitable solvent, filtered using a Pasteur pipette filled with Celite (6cm pad) and analyzed by GC using n-hexadecane as an internal standard. Control experiments showed that the position of the vial inside the autoclave is not influential. The same outcome was obtained when the reaction was repeated by moving a vial from a peripheral to a central position.
With hydrazine hydrate; In ethanol; at 30℃; for 2.5h;Autoclave;
General procedure: The catalytic reduction of nitro aromatics was conducted in a 25 mlTelfon-lined stainless steel autoclave with magnetic stirring. In a typicalprocess, 6 mmol nitroarene, and desired amounts of reducing agent andsolvents were introduced in the reactor. The autoclave was transferredinto a water bath at the set temperature with an accuracy of better than0.2 C for ?0.5 h. Then, 10 mg catalyst was added into the reactionmixture and started the reduction at a stirring rate of 450 rpm. After thereaction, the catalyst was rapidly separated by ltration. n-Decane(500 muL) as standard was added into the ltrate and was dried withanhydrous Na 2 SO 4 . The products were analyzed by gas chromato-graphy-mass spectrometry (GC-MS) (Shimadzu GCMS-QP2010 Plus)and GC (Varian CP-3800) with a capillary column (column VF-1 ms,15 m, 0.25 mm, 0.25 mum) and a ame ionization detector (FID).
2.2.1. 2-Bromo-7-nitro-9H-fluorene (1b)
To a stirred solution of 2-nitrofluorene [41] (1a, 5 g, 23.67 mmol)in dry CH2Cl2 (30 mL) was added Br2 (47.34 mmol) slowly. Themixture was stirred for 5-6 h. The yellow precipitate was filteredoff and washed with 5% NaHSO3. The crude yellowish product was purified by recrystallization from DMF to afford 1b (5.5 g, 80% yield) as light yellow powder.
With chromium(VI) oxide; orthoperiodic acid In acetonitrile at 20℃; for 1h;
99%
With oxygen In N,N-dimethyl acetamide at 0.25℃; for 24h; Schlenk technique;
98%
With air; graphene-supported KOH composite In N,N-dimethyl-formamide at 20℃; for 8h;
98%
With tert.-butylhydroperoxide; 1-butyl-3-methyl-1H-imidazol-3-ium bromide In lithium hydroxide monohydrate at 55℃; for 20h;
General procedure for oxygenation of benzylic C-H bonds
General procedure: Charged 0.5 mmol of 3-(9H-fluoren-2-yl)prop-2-yn-1-ol 13a (110 mg, 0.5 mmol), 0.5 mL of [bmim]Br and TBHP (0.5 mL, 3.6 mmol) were taken in a round bottom flask and heated to 55 °C until completion of the reaction. The reaction course was monitored by TLC. After completion of the reaction, the reaction mixture was extracted using ethyl acetate (3 * 4 mL). The organic layer was concentrated under vacuum and purified by column chromatography on silica gel to afford the desired product as 2-(3-hydroxyprop-1-yn-1-yl)-9H-fluoren-9-one 13b Yellow Solid;
95%
With oxygen; potassium hydroxide In tetrahydrofuran at 20℃; for 12h;
91%
With tert.-butylhydroperoxide In lithium hydroxide monohydrate at 100℃; for 24h;
General procedure for the oxidative reaction
General procedure: Caution. tert-Butyl hydroperoxide is an exceptionally dangerous chemical that is highly reactive, flammable, and toxic. It is corrosive to skin and mucous membranes and causes respiratory distress when inhaled. A solution of secondary alcohol (1 mmol) and 70% TBHP (6 or 10 equiv.) was stirred at 100 °C for 24 h. The reaction mixture was quenched with the saturated solution of sodium thiosulfate (5 mL) and extracted with dichloromethane (3 x 10 mL). The combined dichloromethane extracts were dried over anhydrous Na2SO4 and filtered, and then the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with PE or PE/EtOAc as the eluent to obtain the desired products.
83%
With oxygen; lithium hexamethyldisilazane In tetrahydrofuran at 60℃; for 12h; Sealed tube; Green chemistry; chemoselective reaction;
3.2. Representative Procedure for the Oxidation of Diarylmethane
General procedure: To an 8 mL oven-dried vial, 4-benzylpyridine (0.1 mmol), dry THF (1 mL), LiHMDS (0.15mmol) were added subsequently. The reaction system was sealed by a rubber septum with a needleconnected with O2 balloon. After stirring at 60 °C for 12 h, the reaction mixture was passed througha short pad of silica gel and eluted with ethyl acetate (1 mL × 3). The combined organics wereconcentrated under reduced pressure. The residue was purified by flash chromatography to givethe diarylketone 2a as white solid (15.6 mg, 85% yield).
80%
With tert.-butylhydroperoxide; Cu(OAc)2*H2O In lithium hydroxide monohydrate at 80℃; for 8h;
77%
With Shirasagi KL activated carbon; oxygen In 1,3-dimethylbenzene at 120℃; for 95h;
71%
With chromium(VI) oxide; glacial acetic acid at 20℃; for 24h;
5.1 1) Synthesis of intermediate A5-2
A5-1 (5 g, 23.70 mmol) was dissolved in acetic acid (125 mL) Chromium trioxide (9.5 g, 94.79 mmol) was added. The mixture was stirred at room temperature for 24 hours and filtered to give a yellow solid (3.8 g, 71%).
59%
With copper(II) dichloride dihydrate; 4,7-bis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-1,10-phenanthroline; oxygen; sodium hydroxide In lithium hydroxide monohydrate at 50℃; for 16h; Schlenk technique;
59%
With C26H36N2O6; oxygen; cobalt(II) nitrate; potassium hydroxide In lithium hydroxide monohydrate at 90℃; for 20h;
54%
With sodium bismuthate; glacial acetic acid In lithium hydroxide monohydrate; propan-2-one for 42h; Heating / reflux;
41%
With (4s,6s)-2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile; oxygen; tetra-n-butylammonium azide In acetonitrile at 25℃; for 36h; Irradiation;
With perchloric acid; anhydrous sodium perchlorate; pyridinium fluorochromate In lithium hydroxide monohydrate; glacial acetic acid at 30℃;
With perchloric acid; quinolinium dichromate In N,N-dimethyl-formamide at 40℃;
With sodium dichromate; glacial acetic acid
With pyridine; oxygen; N,N,N-trimethylbenzenemethanaminium hydroxide
With pyridine; potassium hydroxide; potassium peroxodisulfate
With aluminum (III) chloride In nitrobenzene at 45 - 65℃; for 1.5h;
1.1 Synthesis of 1-(9,9-H-7-nitrofluorene-2-yl)-ethanone
After 5.0 g of 2-nitrofluorene (1) (23.7 mmol) was dissolved in 100 ml of anhydrous nitrobenzene and 6.31 g of anhydrous aluminum chloride (47.4mmol) was added thereto, the reaction mixture was heated to 45°C, and a solution in which 2.79g of acetyl chloride (35.5mmol) was dissolved in 30ml of anhydrous nitrobenzene was slowly added thereto for 30 minutes, followed by stirring at 65°C for 1 hour. Then the reaction mixture was cooled to room temperature and 70ml of distilled water was added thereto and stirred for about 30 minutes, followed by filtering the product. The obtained solid product was dispersed in 50ml of ether and stirred at room temperature for 30 minutes, followed by filtering and drying, thereby obtaining 5.08g of 1-(9,9-H-7-nitrofluorene-2-yl)-ethanone (2) (84.7%) as a light yellow product. 1H NMR(δ ppm; DMSO-d6) : 2.64(3H, s), 4.18(2H, s), 8.06(1H, dd), 8.21-8.32(4H, m), 8.51(1H, d) MS(m/e):253
With aluminium trichloride; nitrobenzene at 40 - 55℃;
3,3'-(2-nitro-fluorene-9,9-diyl)-di-propionitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 1,4-dioxane; N-benzyl-trimethylammonium hydroxide
70 g
Stage #1: 2-nitro-9H-fluorene With N-benzyl-trimethylammonium hydroxide In 1,4-dioxane for 0.0833333h;
Stage #2: acrylonitrile In 1,4-dioxane at 40℃;
2 Example 2: Preparation of compound No 2
A mixture of compound 1 (56.4 g) in dioxane (400 mL) was mechanically stirred in a 3 -neck round- bottom flask. The 40% Triton B solution (1.3 mL) was added and the reaction turned deep purple. After 5 minutes of stirring, acrylonitrile (37 mL) was added dropwise. The temperature rose to ~40°C. The reaction was stirred overnight. The reaction was checked by TLC (silica, 1/9, EtOAc/chloroform) and showed complete conversion of the starting material to a lower Rf spot. After the reaction was complete the reaction was concentrated. The solid product was recrystallized from organic solvent to give pure compound 2 (70 g).
70 g
Stage #1: 2-nitro-9H-fluorene With N-benzyl-trimethylammonium hydroxide In 1,4-dioxane for 0.0833333h;
Stage #2: acrylonitrile In 1,4-dioxane at 40℃;
2 Example 2: Preparation of compound No 2
A mixture of compound 1 (56.4 g) in dioxane (400 mL) was mechanically stirred in a 3 -neck round- bottom flask. The 40% Triton B solution (1.3 mL) was added and the reaction turned deep purple. After 5 minutes of stirring, acrylonitrile (37 mL) was added dropwise. The temperature rose to ~40°C. The reaction was stirred overnight. The reaction was checked by TLC (silica, 1/9, EtOAc/chloroform) and showed complete conversion of the starting material to a lower Rf spot. After the reaction was complete the reaction was concentrated. The solid product was recrystallized from organic solvent to give pure compound 2 (70 g).
2-Nitro-9,9-Bis(4-pyridinylmethyl)fluorene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sodium hydroxide In water; toluene
2-Nitro-9,9-Bis(4-pyridinylmethyl)fluorene
2-Nitro-9,9-Bis(4-pyridinylmethyl)fluorene A quantity of 0.5 g (2.37 mmole) of 2-nitrofluorene, 0.86 g (5.2 mmole) of 4-picolylchloride hydrochloride, 60 mg of cetyl tri-n-butyl phosphonium bromide and 10 ml of toluene were combined and heated to 50°. With vigorous stirring 5.0 ml of 50% sodium hydroxide was added dropwise at 50° during 30 minutes. Heating was continued for one hour. A quantity of 10 ml of water was added, the reaction cooled to room temperature and partitioned with methylene chloride. The combined organic layer was extracted with 3*25 ml of 0.5N HCl and the combined aqueous extracts basified with sodium hydroxide. The precipitated product was chromatographed (silica, methylene chloride/methanol 100:1) to yield 0.6 g of the title product, m.p. 260°-264°. HRMS-measured, 393.1465; calculated, 393.1477; assigned, C25 H19 N3 O2 (M+). The dialkylated fluorene was converted to its dihydrochloride salt by dissolving 0.5 g of the base in ethanol and adding 2 ml of 25% hydrochloric acid in ethanol. Addition of ether produced product, which was recrystallized from methanol/ethyl acetate to yield b 0.4 g, m.p. >300°.
{(η5-C5H5)Fe(η5-C5H4CH(2-nitro-9-fluorenyl))}[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
20%
With KOtBu In tetrahydrofuran (Schlenk techniques); the fluorene is added to the carboxaldehyde in THF at room temp., KOtBu is added, stirred for 15 min; diluted with ether, chromy.;
With tetrabutylammomium bromide; sodium hydroxide In water; toluene at 60℃; for 8h; Inert atmosphere;
90%
With [benzene-1,3,5-triyltris(methylene)]tris(triphenylphosphonium) tribromide; sodium hydroxide In water; dimethyl sulfoxide at 30℃; for 6h; Inert atmosphere;
Synthesis of 2-nitro-9,9-dioctyl-9H-fluorene (6a)
To a stirred solution of 2-nitrofluorene 4a (1 g, 4.7 mmol) in DMSO, 25 % aq. sodium hydroxide solution added dropwise over a period of 10 min under nitrogen gas purging, then the catalyst 3 (10 mol %) was added, followed by 1-bromooctane 5a (1.82 g, 9.4 mmol), then the reaction mass was stirred at room temperature. Reaction completion was confirmed by TLC, then extracted with ethyl acetate, washed with water, dried over sodium sulfate and evaporated under reduced pressure, to produce a pale yellow viscous liquid with 90 % yield. 1H NMR (300 MHz, CDCl3) d 8.26 (d, J = 6 Hz, 1H), 8.21 (s, 1H), 7.79 (d, J = 6 Hz, 2H),7.41 (s, 3H), 2.02 (t, J = 6 Hz, 4H), 1.20 - 1.03 (m, 24H), 0.81 (t, J = 4.5 Hz, 6H);13C NMR (75 MHz, CDCl3) d: 152.33, 151.98, 147.67, 147.14, 138.74, 129.28,127.39, 123.28, 123.22, 121.21, 119.79, 118.25, 55.69, 40.06, 31.74, 29.87, 29.18,29.16, 23.74, 22.58, 14.06.
75%
With tetrabutylammomium bromide; sodium hydroxide In water; toluene at 90℃; for 8h; Inert atmosphere;
1 Preparation of 2-nitro-9,9-dialkylfluorene
2-nitroguanidine (10.6 g, 50 mmol) was added to the reaction flask under an argon atmosphere.N-octyl bromide (24.1 g, 125 mmol) at a concentration of 50% by weightAqueous sodium hydroxide solution (10g/10ml deionized water, 0.25mol)Tetrabutylammonium bromide (0.81 g, 2.5 mmol) and 200 ml of toluene solvent,Heat to 90 ° C and react for 8 hours. After stopping the reaction, the organic phase is separated,Concentration, purification by silica gel column chromatography, petroleum ether / dichloromethane mixed solvent (10/1, v / v) as a rinse, a pale yellow liquid,The yield was 75%.
70%
With potassium hydroxide In dimethyl sulfoxide at 20℃; for 1h; Inert atmosphere;
70%
Stage #1: 2-nitro-9H-fluorene With potassium hydroxide In dimethyl sulfoxide for 0.333333h;
Stage #2: 1-bromo-octane In dimethyl sulfoxide for 0.5h;
1 Synthesis of Compound A:
Add 250 mL to a 250 mL flask1,2-dichloroethane, and then 20 mL of concentrated nitric acid (65%) was added to disperse all The reaction was carried out in an ice-water bath followed by the slow addition of 10 g of fluorene. After 20 min reaction, the mixture was poured into 300 mL of ice methanol and filtered to give pale yellow solid 2 (11 g).A solution of 7.5 g of KOH was added to 100 mL of DMSO and dispersed uniformly After the addition of the above pale yellow solid 6.3 g, stirring for 20 min, 15 mL of 1-bromooctane was added dropwise to the above system, The reaction was carried out for 30 min, extracted with petroleum ether, dried and dried to give the crude product, which was then subjected to column chromatography using petroleum ether as developing solvent A yellow viscous liquid, compound A (9.0 g) was obtained.
With triethylamine In dichloromethane at 35℃; for 5h;
1.1 Example 1
1) In a dry 2000 ml four-neck flask, add 2-nitrofluorene 422 g (2.0 mol),Triethylamine (303 g, 3.0 mol) and dichloromethane 800g,301 g (2.2 mol) of bromobutane was added dropwise with stirring (20° C.), and was dripped for about 1 h. temperature rise to 35°C ±2°C for 4hThe solid (triethylamine salt) was filtered off, 500 g of water was added, and the layers were separated. The aqueous phase was combined once with 500 ml of methylene chloride, washed once with 300 ml of water, and the dichloromethane was recovered to dryness. Then 800 g of methanol was used, and the solution was heated to dissolve and then cooled to 0. °C filtration, drying, compound 1 613.7g, yield 95%, content 98.5%.
91%
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 16h; Inert atmosphere;
79.5%
Stage #1: 2-nitro-9H-fluorene With potassium iodide; potassium hydroxide In dimethyl sulfoxide at 15℃; Inert atmosphere;
Stage #2: 1-bromo-butane In dimethyl sulfoxide at 15℃; for 3h; Inert atmosphere;
2.1 Synthesis of 9.9-di-n-butyl-2-nitrofluorene
12.66g of 2-nitrofluorene (1) (60mmol), 21.0g of potassium hydroxide (0.3mol, purity: 80%), and 1.01g of potassium iodide (6mmol) were dissolved in 200ml of anhydrous dimethylsulfoxide under nitrogen atmosphere, and a reaction temperature was maintained at 15°C. Then, 33ml of n-bromobutane (0.3mol) was slowly added thereto for 2 hours, followed by stirring at 15°C for 1 hour. Thereafter, 200ml of distilled water was added to the reaction mixture and stirred for about 30 minutes, a product was extracted with 300ml of dichloromethane, and the extracted organic layer was washed with 100ml of distilled water three times. Then, the recovered organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled under reduced pressure. The obtained product was purified using a silica gel column chromatography (eluent: dichloromethane: n-hexane=20:1), thereby obtaining 15.4g of 9,9-di-n-butyl-2-nitrofluorene (5) (79.5%) as a light yellow product. 1H NMR(δ ppm; CDCl3) : 0.52-0.61(4H, m), 0.66(6H, t), 1.07(4H, sex), 2.00-2.06(4H, m), 7.38-7.42(3H, m), 7.77-7.80(2H, d), 8.20(1H, d),8.26(1H, dd) MS(m/e):323
Stage #1: 2-nitro-9H-fluorene With N-Bromosuccinimide; dibenzoyl peroxide In benzene for 4h; Reflux;
Stage #2: triphenylphosphine In benzene for 48h; Reflux;
Stage #1: 2-nitro-9H-fluorene With N-Bromosuccinimide In benzene for 4h; Heating;
Stage #2: triphenylphosphine In benzene for 48h; Heating;
3-((2-Nitro-9H-fluoren-9-ylidene)methyl)pyridin-2(lH)-one (CYD- 1 -93).
To a solution of 2-nitrofluorene (326 mg, 1.54 mmol) and 2-oxo-l,2-dihydro-pyridine- 3-carbaldehyde (19 mg, 1.54 mmol) in 10 mL of methanol was added KF-AI2O3 (224 mg, 1.38 mmol). The resulting mixture was stirred at 85 °C. After 24 hrs, TLC indicated that a new product was produced and lots of starting material was still remained. 40 mL of CH2C12 was added into the reaction mixture. The insoluble solid was filtrated, and the filtrate was concentrated under vacuum to give a yellow solid, which was purified by silica gel column; eluting with 60% EtOAc in hexane afforded 26 mg of CYD- 1-93 as a yellow solid. 1H-NMR (600 MHz, d6-DMSO) δ 12.18 (br s, 2H), 8.77 (d, 1H, J = 1.8 Hz), 8.52 (d, 1H, J = 1.8 Hz), 8.28 (m, 2H), 8.15 (m, 2H), 8.10 (d, 1H, J = 7.8 Hz), 8.06 (m, 2H), 7.97 (m, 2H), 7.86 (m, 2H), 7.80 (s, 1H), 7.63 (m, 1H), 7.59 (m, 1H), 7.49 (m, 3H), 7.37 (m, 1H), 6.40 (m, 2H). 13C-NMR (150 MHz, CDCI3) δ 161.2, 161.1, 146.9, 146.2, 146.1, 143.6, 142.1, 141.7, 140.5, 139.6, 138.3, 137.3, 136.1, 136.0, 133.6, 133.4, 129.3, 129.1, 129.0, 128.8, 127.9, 127.4, 126.1, 125.9, 124.0, 123.9, 123.5, 121.9, 121.5, 121.1, 120.7, 120.5, 119.0, 105.1, 105.0.
2-[3-(2-Nitro-fluoren-9-ylidenemethyl)-pyridin-2-yloxy]-ethylamine (CYD-4-61)
To a solution of 2-fluoro-pyridine-3-carbaldehyde (500 mg, 3.995 mmol) and (2- hydroxy-ethyl)-carbamic acid tert-butyl ester (1287 mg, 7.99 mmol) in 20 mL of DMF was added Na2C03 (847 mg, 7.99 mmol). The resulting mixture was stirred at 80 °C for 5 hrs and the reaction progress was monitored by TLC analysis. The reaction mixture was then washed with brine, and concentrated under vacuum to give an oil residue, which was purified by silica gel column; eluting with EtOAc/hexane = 1 :2 to afford 600 mg of CYD-5-75 in 60% yield as colorless gel. To a solution of 2-nitrofluorene (244 mg, 1.15 mmol) and CYD-5-75 (220 mg, 0.82 mmol) in 20 mL of methanol was added KF-A1203 (184 mg, 1.15 mmol). The resulting mixture was stirred at 72 °C. After 6 hrs, TLC indicated that the starting material was gone. 40 mL of CH2C12 was added into the reaction mixture. The insoluble solid was filtrated, and the filtrate was concentrated under vacuum to give a yellow solid, which was recrystallized from alcohol and CH2C12 to give 120 mg of a yellow solid. The yelow solid (180 mg, 0.39 mmol) was dissolved in 4 mL of CH2C12, and then lmL of TFA was added into it at 0 °C. The resulting mixture was stirred at rt for 4 hrs. The reaction mixture was washed with sat. NaHC03 (aq.), and concentrated under vacuum to give an oil residue, which was purified by silica gel column; eluting with CH2Cl2/MeOH = 20: 1 to provide 150 mg of CYD-4-61 as yellow soild in 50% yield for two steps . 1H-NMR (600 MHz, CDC13) δ 8.61 (d, 1H, J= 1.8 Hz), 8.40 (d, 1H, J = 2.4 Hz), 8.27 (m, 2H), 8.23 (dd, 1H, J= 2.4 Hz, 8.4 Hz), 8.18 (dd, 1H, J= 1.8 Hz, 7.8 Hz), 7.89 (m, 3H), 7.76 (m, 4H), 7.72 (s, 1H), 7.68 (s, 1H), 7.61 (d, 1H, J= 7.8 Hz), 7.45 (m, 2H), 7.39 (m, 1H), 7.23 (m, 1H), 7.04 (m, 1H), 7.00 (m, 1H), 4.46 (m, 4H), 3.07 (m, 4H), 1.45 (br s, 4H). 13C-NMR (150 MHz, CDC13) δ 161.3, 161.2, 148.0, 147.7, 147.1, 146.6 (2C), 144.4, 140.7, 139.9, 139.5, 139.4, 138.9, 137.8, 136.8, 136.6, 135.5, 135.4, 129.2, 129.0, 128.9, 128.7, 124.6, 124.5, 124.3, 124.0, 123.7, 121.2, 120.9, 119.7, 119.6, 119.4, 118.7, 118.4, 116.7, 116.6, 116.1, 68.8, 68.7, 41.3 (2C). HRMS calc. for C2iHi7N303 [M+H]+ 360.1343; found 360.1351. HPLC purity 99.2%
With potassium fluoride on basic alumina In methanol at 65℃; for 16h;
General procedure: To a solution of 2-nitrofluorene (63 mg, 0.3 mmol) and 39a(80 mg, 0.3 mmol) in 5mL of methanolwas added KF-Al2O3 (48 mg,0.3 mmol). The reaction mixture was stirred at 65 °C for 16 h, andthen evaporated in vacuo to provide 105mg of crude product 40a.40a was then dissolved in 3mL of CH2Cl2. TFA (456 mg, 4.0 mmol)was added and the mixture was stirred overnight at rt. Then thereactionwas adjusted to pH 8 by adding saturated NaHCO3 and wasextracted with CH2Cl2 (3 x 10 mL). The combined organic layer waswashed withwater, dried over Na2SO4, filtered, and concentrated invacuo. The residue was purified by silica column chromatography(MeOH/CH2Cl2, 1:10) to give the final compound 41a as a yellowfoam.
With potassium fluoride on basic alumina In methanol at 70℃;
4.1.1. 2-((3-((4H-Indeno[1,2-b]thiophen-4-ylidene)methyl)pyridin-2-yl)oxy)ethan-1-amine (5)
General procedure: Thiophen-2-ylboronic acid (256 mg, 2.0 mmol) and Pd(PPh3)4(116 mg, 0.1 mmol) were added to an oven-dried Schlenk flask.After degassed with N2, 1-bromo-2-iodobenzene (622 mg,2.2 mmol), toluene (8 mL), ethanol (4 mL) and 1MNa2CO3 aqueoussolution (4 mL) were added and the reaction mixture was stirred at80 °C overnight. After cooled to room temperature, 10 mL of Et2Owas added and the organic layer was then washed with saturatedbrine (10 mL) and dried over anhydrous Na2SO4. The solvent wasevaporated in vacuo and the resulting residue was purified by silicagel chromatography to give compound 2 as a colorless oil. Yield255 mg, 54%. Compound 2 (172 mg, 1.0 mmol), Pd(PPh3)4 (57 mg,0.05 mol), K2CO3 (138 mg, 1.0 mmol) and KOAc (98 mg, 1.0 mmol)were mixed in 10 mL of dioxane. After degassed with N2, trimethylsilyldiazomethane (2 N in hexane, 0.6 mL,1.2 mmol)was addedvia syringe. The mixture was stirred at 100 °C overnight and thenfiltered through celite. The solvents were evaporated in vacuo andthe residue was purified by silica gel chromatography to givecompound 4 as a pale solid. Yield 82 mg, 48%. To a solution ofcompound 4 (35 mg, 0.2 mmol) and tert-butyl (2-((3-formylpyridin-2-yl)oxy)ethyl)carbamate (53 mg, 0.2 mmol) in10 mL of methanol was added KF-Al2O3 (39 mg, 0.24 mmol). Thereaction mixture was stirred at 70 °C overnight, and then evaporatedin vacuo. The residue was purified by silica gel columnchromatography (ethyl acetate/CH2Cl2, 1:4) to give an intermediate,which was dissolved in CH2Cl2 (5 mL) and TFA (228 mg, 2.0 mmol)was slowly added. The reactionwas stirred at rt overnight and thentreated with saturated NaHCO3 (5 mL). The mixture was extractedwith CH2Cl2 (3 10 mL), dried over Na2SO4, filtered, concentratedand purified by silica gel column chromatography (CH2Cl2/MeOH)to give compound 5 as a pale yellow foam. Yield 30 mg, 47% (two steps).
With potassium fluoride on basic alumina; In methanol; at 65℃; for 16h;
General procedure: To a solution of 2-nitrofluorene (211 mg, 1.0 mmol) and o-anisaldehyde(272 mg, 2.0 mmol) in 5mL of methanol was added KFAl2O3(160 mg, 1.0 mmol). The reaction mixture was stirred at 65 Cfor 16 h, and then evaporated in vacuo. The residue was purified bysilica gel column chromatography (ethyl acetate/CH2Cl2, 1:2) toprovide compound 6 as a yellow foam.
306mg
With KF-Al2O3; In methanol; at 72℃; for 8h;
To a solution of 2-nitrofluorene (278 mg, 1.32 mmol) and 2-methoxy-3- pyridinecarboxyaldehyde (200 mg, 1.46 mmol) in 15 mL of methanol was added KF-AI2O3 (190 mg, 1.18 mmol). The resulting mixture was stirred at 72 C. After 8 hrs, TLC indicated that the starting material was gone. 40 mL of CH2CI2 was added into the reaction mixture. The insoluble solid was filtrated, and the filtrate was concentrated under vacuum to give a yellow solid, which was recrystallized from alcohol and CH2CI2 to give 306 mg of CYD-1-76 as a yellow solid. 1H-NMR (600 MHz, CDC13) delta 8.88 (s, 1H), 8.32 (m, 10H), 8.10 (m, 22H), 8.01 (d, 1H, J = 7.2 Hz), 7.94 (s, 3H), 7.53 (m, 7H), 7.48 (m, 1H), 7.43 (d, 1H, J = 7.8 Hz), 7.31 (m, 1H), 7.19 (m, 4H), 3.94 (s, 14H).
9-(2-Methoxy-benzylidene)-2-nitro-9H-fluorene (CYD- 1-70).
To a solution of 2-nitrofluorene (1.05 g, 5 mmol) and 2-methoxybenzaldehyde (0.816 g, 6 mmol) in 20 mL of methanol was added KF-AI2O3 (0.75 g, 4.5 mmol). The resulting mixture was stirred at 72 °C. After 6 hrs, TLC indicated that the starting material was gone. 40 mL of CH2CI2 was added into the reaction mixture. The insoluble solid was filtrated, and the filtrate was concentrated under vacuum to give a yellow solid, which was recrystallized from alcohol and CH2CI2 to give 1.2 g of CYD-1-70 as a yellow solid.1H-NMR (600 MHz, CDC13) δ 8.84 (s, 0.36H), 8.26 (m, 1.65H), 8.10 (m, 3.05H), 8.02 (s, 0.57H), 7.55 (m, 4H), 7.24 (m, 1.24H), 7.10 (m, 0.91H), 3.84 (s, 3H). 13C-NMR (150 MHz, CDC13) δ 157.7, 147.4, 146.6, 146.5, 144.2, 140.8, 140.0, 138.6, 137.9, 136.8, 136.6, 134.2, 134.0, 131.5, 131.2, 131.1, 130.8, 129.6, 129.5, 129.2 (2C), 128.9, 128.4, 124.5, 124.3, 124.2, 124.1, 123.9, 122.2, 121.9, 121.6, 121.1, 120.9, 120.8 (2C), 119.1, 116.6 (2C), 112.1, 112.0, 55.9 (2C).
With potassium fluoride on basic alumina In methanol
125mg
With KF-Al2O3 In methanol at 72℃; for 6h;
2-(2-Nitro-fluoren-9-ylidenemethyl)-phenol (CYD- 1 -87).
To a solution of 2-nitrofluorene (250 mg, 1.18 mmol) and salicylaldehyde (159 mg, 1.30 mmol) in 10 mL of methanol was added KF-AI2O3 (170 mg, 1.06 mmol). The resulting mixture was stirred at 72 °C. After 6 hrs, TLC indicated that the starting material was gone. 40 mL of CH2CI2 was added into the reaction mixture. The insoluble solid was filtrated, and the filtrate was concentrated under vacuum to give a yellow solid, which was purified by silica gel column; eluting with 11% EtOAc in hexane afforded 125 mg of CYD-1-87 as a yellow solid.1H-NMR (600 MHz, CDC13) δ 10.05 (br s, 1H), 8.39 (s, 1H), 8.27 (dd, 1H, J = 2.4 Hz, 8.4 Hz), 8.16 (d, 1H, J = 8.4 Hz), 8.09 (m, 2H), 8.04 (s, 1H), 7.52 (m, 3H), 7.37 (m, 1H), 7.05 (d, 1H, J = 8.4 Hz), 6.96 (t, 1H, J = 7.2 Hz).
1-(9,9-di-n-butyl-7-nitrofluorene-2-yl)-ethanone oxime-O-acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium hydroxide In water; dimethyl sulfoxide
2 Reaction 1.
Preparation of 1-(9,9-di-n-butyl-7-nitrofluorene-2-yl)-ethanone oxime-O-acetate Reaction 1. Synthesis of 9.9-di-n-butyl-2-nitrofluorene 12.66 g of 2-nitrofluorene (1) (60 mmol), 21.0 g of potassium hydroxide (0.3 mol, purity: 80%), and 1.01 g of potassium iodide (6 mmol) were dissolved in 200 ml of anhydrous dimethylsulfoxide under nitrogen atmosphere, and a reaction temperature was maintained at 15° C. Then, 33 ml of n-bromobutane (0.3 mol) was slowly added thereto for 2 hours, followed by stirring at 15° C. for 1 hour. Thereafter, 200 ml of distilled water was added to the reaction mixture and stirred for about 30 minutes, a product was extracted with 300 ml of dichloromethane, and the extracted organic layer was washed with 100 ml of distilled water three times. Then, the recovered organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled under reduced pressure. The obtained product was purified using a silica gel column chromatography (eluent: dichloromethane:n-hexane=20:1), thereby obtaining 15.4 g of 9,9-di-n-butyl-2-nitrofluorene (5) (79.5%) as a light yellow product. 1H NMR (δ ppm; CDCl3): 0.52-0.61 (4H, m), 0.66 (6H, t), 1.07 (4H, sex), 2.00-2.06 (4H, m), 7.38-7.42 (3H, m), 7.77-7.80 (2H, d), 8.20 (1H, d), 8.26 (1H, dd)
With manganese; (1,2-dimethoxyethane)dichloronickel(II); 4,4'-Dimethoxy-2,2'-bipyridin; trimethylsilyl iodide In N,N-dimethyl-formamide at 120℃; for 16h;
With aluminum (III) chloride In dichloromethane at 0 - 10℃; for 4h;
2.1 (1) Preparation of Intermediate 2a
To a 500 mL four-necked flask, 105 g of starting material 2a, 67 g of aluminum chloride, and 100 mL of dichloromethane were added. The ice water bath was lowered to 0° C., and a mixed solution of 80 g of the starting material 2b and 50 mL of dichloromethane was added dropwise to the temperature of 10° C. or lower. After about 2 hours, the mixture was added dropwise. After stirring was continued for 2 hours, the liquid phase was traced to completion. The material was slowly poured into dilute hydrochloric acid (800 g of ice water and 100 mL of concentrated hydrochloric acid (37%)) and stirred while stirring. Pour into a separatory funnel, separate the lower methylene chloride layer, and continue to wash the aqueous layer with 50 mL of methylene chloride. Combine the methylene chloride layers and wash them with 5% aqueous sodium bicarbonate (300 mL each time, 3 times in total). Chloroform layer, followed by washing the dichloromethane layer until the pH is neutral, drying the dichloromethane layer with 150g of anhydrous magnesium sulfate, filtering and vortexing the dichloromethane solution, recrystallizing from methanol, oven drying at 70°C for 2 hours to obtain 132g Formula 2a, yield 79%, purity 98%.
With tungsten hexacarbonyl; triphenylphosphine at 22℃; for 24h; Inert atmosphere; Schlenk technique; Sealed tube; Irradiation;
76%
Stage #1: 2-Nitrofluorene With [2,2]bipyridinyl; MoO<SUB>2</SUB>Cl<SUB>2</SUB>(DMF)<SUB>2</SUB> In toluene for 0.0333333h;
Stage #2: phenylboronic acid With triphenylphosphine In toluene at 100℃; for 14h;
Stage #1: 2-nitro-9H-fluorene; benzoic acid ethyl ester With chromium chloride; chloro-trimethyl-silane; magnesium; 4,4'-di-tert-butyl-2,2'-bipyridine In tetrahydrofuran at 90℃; for 12h; Schlenk technique; Inert atmosphere;
Stage #2: With hydrogenchloride; water In tetrahydrofuran
(E)-3-((2-nitro-9H-fluoren-9-ylidene)methyl)pyridin-2-ol[ No CAS ]
(Z)-3-((2-nitro-9H-fluoren-9-ylidene)methyl)pyridin-2-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium fluoride on basic alumina; In methanol; at 65℃; for 16h;
General procedure: To a solution of 2-nitrofluorene (211 mg, 1.0 mmol) and o-anisaldehyde(272 mg, 2.0 mmol) in 5mL of methanol was added KFAl2O3(160 mg, 1.0 mmol). The reaction mixture was stirred at 65 Cfor 16 h, and then evaporated in vacuo. The residue was purified bysilica gel column chromatography (ethyl acetate/CH2Cl2, 1:2) toprovide compound 6 as a yellow foam.
1-(3-((2-nitro-9H-fluoren-9-ylidene)methyl)pyridin-2-yl)piperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61%
With potassium fluoride on basic alumina In methanol at 65℃; for 16h;
4.1.4. (E/Z)-9-(2-methoxybenzylidene)-2-nitro-9H-fluorene (6)
General procedure: To a solution of 2-nitrofluorene (211 mg, 1.0 mmol) and o-anisaldehyde(272 mg, 2.0 mmol) in 5mL of methanol was added KFAl2O3(160 mg, 1.0 mmol). The reaction mixture was stirred at 65 °Cfor 16 h, and then evaporated in vacuo. The residue was purified bysilica gel column chromatography (ethyl acetate/CH2Cl2, 1:2) toprovide compound 6 as a yellow foam.
(E)-1-methyl-4-(3-((2-nitro-9H-fluoren-9-ylidene)methyl)pyridin-2-yl)piperazine[ No CAS ]
(Z)-1-methyl-4-(3-((2-nitro-9H-fluoren-9-ylidene)methyl)pyridin-2-yl)piperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
10 % de
With potassium fluoride on basic alumina In methanol at 65℃; for 16h; Overall yield = 73percent; Overall yield = 70 mg;
4.1.4. (E/Z)-9-(2-methoxybenzylidene)-2-nitro-9H-fluorene (6)
General procedure: To a solution of 2-nitrofluorene (211 mg, 1.0 mmol) and o-anisaldehyde(272 mg, 2.0 mmol) in 5mL of methanol was added KFAl2O3(160 mg, 1.0 mmol). The reaction mixture was stirred at 65 °Cfor 16 h, and then evaporated in vacuo. The residue was purified bysilica gel column chromatography (ethyl acetate/CH2Cl2, 1:2) toprovide compound 6 as a yellow foam.
With potassium fluoride on basic alumina In methanol at 65℃; for 16h;
To a solution of 2-nitrofluorene (63 mg, 0.3 mmol) and 39a(80 mg, 0.3 mmol) in 5mL of methanolwas added KF-Al2O3 (48 mg,0.3 mmol). The reaction mixture was stirred at 65 °C for 16 h, andthen evaporated in vacuo to provide 105mg of crude product 40a.40a was then dissolved in 3mL of CH2Cl2. TFA (456 mg, 4.0 mmol)was added and the mixture was stirred overnight at rt. Then thereactionwas adjusted to pH 8 by adding saturated NaHCO3 and wasextracted with CH2Cl2 (3 x 10 mL). The combined organic layer waswashed withwater, dried over Na2SO4, filtered, and concentrated invacuo. The residue was purified by silica column chromatography(MeOH/CH2Cl2, 1:10) to give the final compound 41a as a yellowfoam.
With potassium fluoride on basic alumina In methanol at 65℃; for 16h;
General procedure: To a solution of 2-nitrofluorene (63 mg, 0.3 mmol) and 39a(80 mg, 0.3 mmol) in 5mL of methanolwas added KF-Al2O3 (48 mg,0.3 mmol). The reaction mixture was stirred at 65 °C for 16 h, andthen evaporated in vacuo to provide 105mg of crude product 40a.40a was then dissolved in 3mL of CH2Cl2. TFA (456 mg, 4.0 mmol)was added and the mixture was stirred overnight at rt. Then thereactionwas adjusted to pH 8 by adding saturated NaHCO3 and wasextracted with CH2Cl2 (3 x 10 mL). The combined organic layer waswashed withwater, dried over Na2SO4, filtered, and concentrated invacuo. The residue was purified by silica column chromatography(MeOH/CH2Cl2, 1:10) to give the final compound 41a as a yellowfoam.
N-(9H-fluoren-2-yl)-2-phenylethane-1-sulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
With sodium metabisulfite; lithium phosphate; choline chloride In N,N-dimethyl-formamide at 130℃; for 10h; Inert atmosphere;
33 Example 33Synthesis of compound 35
Add 2-nitrofluorene (0.2mmol), sodium metabisulfite (0.6mmol, 3.0equiv), phenethylboronic acid (0.3mmol, 1.5equiv) to the reaction tube,Choline chloride (0.2 mmol, 1.0 equiv), lithium phosphate (0.4 mmol, 2.0 equiv),Evacuate and change nitrogen three times and add solvent DMF (2.0mL),The reaction system was heated to 130°C for 10 hours. After all the nitro compounds were completely converted, the reaction system was cooled to room temperature, quenched by adding water, and extracted with ethyl acetate (10 mL*3),Dry with anhydrous sodium sulfate, filter, concentrate,Column chromatography was separated to obtain the purified target product 35 (60%).
Stage #1: 2-nitro-9H-fluorene With caesium carbonate In N,N-dimethyl-formamide at 0℃; for 0.5h; Inert atmosphere;
Stage #2: propyl bromide In N,N-dimethyl-formamide at 20℃; Inert atmosphere;
3.1 Step 1: Preparation of 2-nitro-9,9-dipropyl-9H-fluorene
DMF (50 mL) containing 2-nitro-9H-fluorene (3 g, 14.2 mmol) and Cs2CO3 (13.9 g, 42.6 mmol) was stirred at 0° C. for 30 minutes in argon atmosphere. Bromopropane (3.10 mL, 34.1 mmol) was added to the reaction mixture, which was stirred overnight at room temperature. The reactant was quenched with water and the water layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Silica gel mesh was prepared from the residue, and flash chromatography (eluent: EtOAc/hexane) was performed to give 9,9-diethyl-2-nitro-9H-fluorene (2.99 g, 71%).
methyl 4-((9H-fluoren-2-yl)amino)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
With (4,4′-di-tert-butyl-2,2′-dipyridyl)Ni(o-tolyl)(Br); N-ethyl-N,N-diisopropylamine In toluene at 70℃; for 12h; Molecular sieve; Inert atmosphere; Sealed tube; Irradiation;
1-iodo-2-(((2-methylallyl)oxy)methyl)benzene[ No CAS ]
[ 199620-15-0 ]
N-(9H-fluoren-2-yl)-2-(4-methylisochroman-4-yl)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With tripotassium phosphate tribasic; water monomer; palladium diacetate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene In 1,4-dioxane at 120℃; for 24h; Sealed tube; Inert atmosphere;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping